
Болезнь крейтцфельдта-якоба на мрт
Обновлено: 24.04.2024
Диагностика болезни Крейтцфельдта-Якоба по КТ, МРТ, ПЭТ головного мозга
а) Терминология:
1. Сокращения:
• Болезнь Крейтцфельдта-Якоба (БКЯ)
• Спорадическая болезнь Крейтцфельдта-Якоба (сБКЯ)
• Вариантная болезнь Крейтцфельдта-Якоба (вБКЯ)
2. Определение:
• Быстро прогрессирующее, смертельное нейродегенеративное заболевание, вызываемое прионами (белковые инфекционные частицы, не содержащие ДНК и РНК):
о Трансмиссивная губчатая энцефалопатия
б) Визуализация:
1. Общие характеристики болезни Крейтцфельдта-Якоба (БКЯ):
• Лучший диагностический критерий:
о Повышение интенсивности сигнала от базальных ганглиев (БГ), таламусов и коры больших полушарий на Т2-ВИ прогрессирующего характера
• Локализация:
о Преимущественное поражение серого вещества (СВ)
- БГ: хвостатые ядра и скорлупа > бледные шары (БШ)
- Таламусы (часто при вБКЯ)
- Кора больших полушарий (наиболее часто в процесс вовлекаются лобные, теменные и височные доли):
Вовлечение коры часто имеет асимметричный характер
Вариант Хайденхайна: затылочные доли
Вариант Браунэлл-Оппенгеймера: мозжечок
о Белое вещество (БВ) обычно не поражается

2. КТ признаки болезни Крейтцфельдта-Якоба (БКЯ):
• Бесконтрастная КТ: обычно нормальная картина:
о При КТ обследованиях в динамике возможно выявление быстропрогрессирующей атрофии и расширения желудочков
4. Радионуклидная диагностика болезни Крейтцфельдта-Якоба (БКЯ):
• ПЭТ: регионарный гипометаболизм глюкозы коррелирует с участками поражения на патоморфологических препаратах
• ОФЭКТ с N-изопропил-n-(I-123)-иодамфетамином
о ↓ поглощения РФП и ↓ абсолютных значений rCBF в различных частях коры больших полушарий
5. Рекомендации по визуализации:
• Лучший инструмент визуализации: МРТ с ДВИ и FLAIR
в) Дифференциальная диагностика болезни Крейтцфельдта-Якоба (БКЯ):
1. Гипоксически-ишемическое поражение:
• Наблюдается вовлечение в процесс БГ и парасагиттальных областей коры
• Гиперинтенсивные очаги поражений в БГ на Т1-ВИ и Т2-ВИ
• ДВИ + симметричное вовлечение в процесс СВ
2. Синдром осмотической демиелинизации:
• Экстрапонтинная локализация: повышение интенсивности сигнала от скорлупы и хвостатых ядер на Т2-ВИ
• В острый период - ограничение диффузии на ДВИ
3. Синдром Лея:
• В первую очередь характерен для детского возраста
• Повышение интенсивности сигнала от скорлупы и БШ на Т2-ВИ
4. Другие причины деменции:
• Болезнь Альцгеймера
• Деменция при болезни двигательного нейрона
• Лобно-височная деменция
• Мультиинфарктная деменция
5. Кортикобазальная дегенерация:
• Потеря нейронов в черной субстанции, коре лобно-теменных отделов больших полушарий и полосатых телах (атрофия БГ может быть выражена незначительно)
• МРТ: симметричная/асимметричная атрофия пре- и постцентральных извилин; выраженное поражение парасагиттальных отделов
• Субкортикальный глиоз: повышение интенсивности сигнала на Т2-ВИ
6. Болезнь Вильсона:
• Поражения БВ и субкортикального СВ (БГ, зубчатые ядра, ствол мозга); вариабельный характер повышения интенсивности сигнала на Т2-ВИ
• Гипоинтенсивные поражения на Т1-ВИ (реже гиперинтенсивные)
7. Атеросклероз:
• Вовлечение в процесс БГ: обычно асимметричный и мультифокальный характер
• (а не диффузный, что характерно для БКЯ)
• Локальные гиперинтенсивные очаги в глубоком БВ
• Отсутствие ограничения диффузии на ДВИ, исключая острую фазу
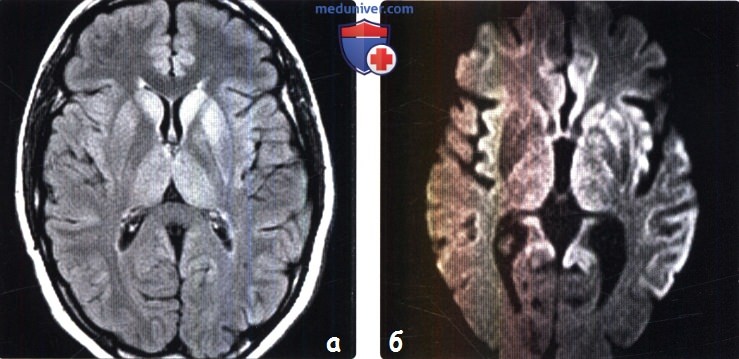
(а) MPT, FLAIR, аксиальный срез: определяется асимметричное повышение интенсивности сигнала от головок хвостатых ядер, больше слева, и левой скорлупы.
(б) МРТ, ДВИ, аксиальный срез: у того же пациента через несколько недель отмечается асимметричное повышение интенсивности сигнала от базальных ганглиев, больше слева, и от коры больших полушарий. При БКЯ более часто наблюдается асимметричное вовлечение в процесс коры, чем базальных ганглиев. У этого пожилого мужчины наблюдалась быстро прогрессирующая деменция и был поставлен диагноз вероятная БКЯ, так как при ЭЭГ были выявлены характерные признаки.
г) Патология:
1. Общие характеристики болезни Крейтцфельдта-Якоба (БКЯ):
• Этиология:
о Прионные белки представляют собой неправильно свернутую изоформу (PrPSc) кодируемого в норме геномом хозяина белка (РгРс)
о PrPSc, введенный в здоровые клетки → запускает самовоспроизводящийся порочный цикл: РгРс → PrPSc
о сБКЯ: спонтанное образование РгРс → PrPSc или вследствие соматической мутации
о Семейная форма БКЯ (семБКЯ): мутации в гене PRNP О БКЯ ятрогенного характера: распространение инфекции из прионсодержащего материала:
- Хирургические инструменты, трансплантаты из твердой мозговой оболочки
- Трансплантация трупной роговицы, препараты гормона роста человека
о вБКЯ: губчатая энцефалопатия крупного рогатого скота передается людям через зараженную говядину:
- также известна как новый вариант БКЯ (нвБКЯ)
• Генетика:
о Может иметь наследственный, спорадический или приобретенный (инфекционный) характер
о 10-15% прионных заболеваний человека связаны с мутациями гена (PRNP) прионного белка (PrPc), локализованного на 20-й хромосоме и имеют доминантно-аутосомный тип наследования
• Ассоциированные аномалии
о ЭЭГ: периодические [высоковольтные] спайк-волна комплексы (ППВК) на фоне низковольтажной активности
2. Макроскопические и хирургические особенности:
• Легкая атрофия коры больших полушарий
• Расширение желудочков
3. Микроскопия:
• Спонгиозная энцефалопатия: наиболее выражено поражение СВ:
о Выраженная потеря нейронов с признаками реактивного астроцитоза
о Заместительный глиоз
о Вакуолизация нейронов со спонгиозными изменениями
• У 10% пациентов с БКЯ наблюдается отложение амилоидных бляшек в мозжечке или полушариях большого мозга
• Вариабельный характер накопления PrPSc в ткани головного мозга
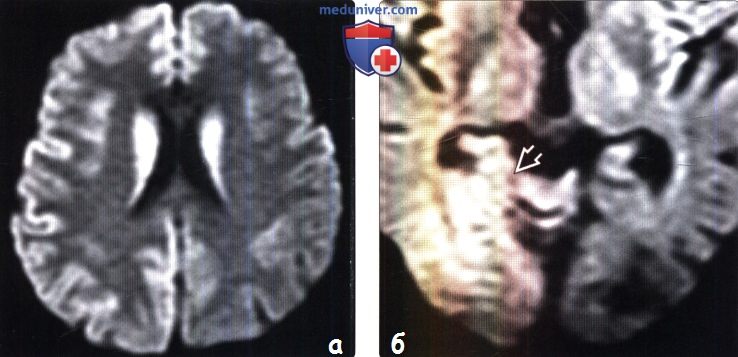
(а) МРТ, ДВИ, аксиальный срез: у данного пациента с БКЯ определяется выраженное повышение интенсивности сигнала от хвостатых ядер и коры вследствие ограничения диффузии. ДВИ является наиболее чувствительным методом для диагностики БКЯ.
(б) МРТ, ДВИ, аксиальный срез: у пациента с начальными жалобами на зрительные нарушения в правой затылочной доле и левом островке визуализируются выражено гиперинтенсивные зоны. При варианте Хайденхайна БКЯ изначально возникают изолированные симптомы зрительных нарушений. При диагностической визуализации отмечается преимущественное вовлечение в процесс лобных долей.
д) Клиническая картина:
1. Проявления болезни Крейтцфельдта-Якоба (БКЯ):
• Наиболее частые признаки/симптомы:
о Прогрессирующая деменция в сочетании с миоклоническими судорогами и акинетическим мутизмом о Вариабельная картина нарушения пирамидных, экстрапирмидных и мозжечковых функций
• Клинический профиль:
о сБКЯ: нарушение функции мозжечка, быстро прогрессирующие когнитивные нарушения или их сочетание:
- 6 молекулярных подтипов: ММ1, ММ2, MV1, MV2, VV1 и VV2:
Варьируют в зависимости от возраста начала заболевания, продолжительности, ранних симптомов и патологических изменений
о вБКЯ: симптомы нарушения психических и чувствительных функций
- Вариант Хайденхайна БКЯ:
Изолированные зрительные признаки/симптомы (первоначально)
Преимущественная дегенерация затылочных долей
- Вариант Браунэлл-Оппенгеймера: мозжечковые признаки/симптомы
- Экстра пирамидный тип БКЯ:
Возможно обнаружение ↑ интенсивности сигнала от БГ
- Вовлечение структур пирамидной системы при прогрессировании заболевания
о Исследования СМЖ:
- Белки -биомаркеры СМЖ: белок 14-3-3, общий тау-белок (о-тау) и нейрон-специфическая энолаза (НСЭ)
- МРТ с ДВИ имеет более высокую точность, чем любые или все три биомаркера СМЖ
о Вибрационно-индуцированный конверсионный анализ СМЖ в режиме реального времени (RT-QUIC) для определения PrPsc:
- Большей чувствительностью обладает исследование обонятельного эпителия (соскоб со слизистой оболочки носа), по сравнению с СМЖ
2. Демография:
• Возраст:
о Молодой при вБКЯ, пожилой при сБКЯ (с 6-й по 7-й декады жизни)
• Этническая принадлежность:
о сБКЯ распространен повсеместно, среди всех рас
о Распространение вБКЯ территориально ограничивается Европой (все случаи отмечаются в Великобритании)
• Эпидемиология:
о В США частота встречаемости 1-1,5 на 1 млн
о сБКЯ (85%), семейные (15%), инфекционные/ятрогенные (менее чем 1 %)формы
3. Течение и прогноз:
• Длительный инкубационный период, но быстрое прогрессирование после появления клинических симптомов
• Быстро прогрессирующая деменция с наступлением летального исхода обычно в течение нескольких месяцев от начала заболевания:
о Медиана выживаемости с момента появления симптомов до смерти составляет 4,5 месяца
о У 90% пациентов продолжительность жизни составляет < 1 года
4. Лечение:
• Эффективное лечение отсутствует
е) Диагностическая памятка:
1. Обратите внимание:
• Предполагайте вариант Хайденхайна БКЯ у пациентов с нарушением зрения неясного происхождения и деменцией
2. Советы по интерпретации изображений:
• Отсутствие признаков поражения БГ не исключает БКЯ
Диагностика прогрессирующего надъядерного паралича по КТ, МРТ, ПЭТ головного мозга
а) Терминология:
1. Сокращения:
• Прогрессирующий надъядерный паралич (ПНП)
2. Синонимы:
• Синдром Стила-Ричардсона-Ольшевского
3. Определение:
• Нейродегенеративное заболевание, характеризующееся надъядерным параличом, постуральной неустойчивостью, легкой деменцией
б) Визуализация:
2. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ
о ПЭТ
• Советы по протоколу исследования:
о Срединные сагиттальные Т1-ВИ
о 3D-изображения в режиме 3D MPRAGE или FSPGR:
- Используйте вексельную морфометрию для расчета отношения площади среднего мозга к площади моста
3. КТ признаки прогрессирующего надъядерного паралича:
• Бесконтрастная КТ:
о Атрофия среднего мозга с выраженным расширением цистерны среднего мозга и III желудочка
в) Дифференциальная диагностика прогрессирующего надъядерного паралича:
1. Множественная системная атрофия, паркинсонический тип (МСА-П):
• Снижение интенсивности сигнала от скорлупы на Т2-ВИ без выраженной атрофии среднего мозга
• Атрофия мозжечка и моста
• Выраженью симптомы нарушения функции мозжечка, вегетативной дисфункции, паркинсонизм
3. Деменция с тельцами Леви:
• Атрофия коры без выраженной атрофии среднего мозга
• Галлюцинации, корковая деменция с афазией, паркинсонизм
4. Болезнь Паркинсона:
• Выраженная атрофия среднего мозга отсутствует
• В случаях, когда ведущим клиническим симптомом является тремор, наблюдается хороший ответ на терапию леводопой
г) Патология:
1. Общие характеристики прогрессирующего надъядерного паралича:
• Этиология:
о ПНП является таупатией
о Аномальное накопление фосфорилированного тау-белка в головном мозге:
- Бледные шары, субталамические ядра, красные ядра, черная субстанция, покрышка моста, полосатые тела, глазодвигательные ядра, продолговатый мозг, зубчатые ядра
• Генетика:
о Связь с тау-белком, ген МТАР в 17 хромосоме
• Гаплотип Н1 тау-белка связан как с РПНП, так и с кортикобазальной дегенерацией:
о Предполагает, что аномалии гена тау-белка на хромосоме 17 являются причинами обеих болезней
2. Макроскопические и хирургические особенности:
• Атрофия субталамических ядер и ствола мозга (покрышки среднего мозга и верхних ножек мозжечка)
• Потеря пигментации черной субстанции → дегенерация нигростриарного дофаминергического пути
3. Микроскопия:
• Потеря нейронов, астроцитарный глиоз
• Нейрофибриллярные клубки и нити в нейропилях в бледных шарах, субталамических ядрах, черной субстанции; кора больших полушарий относительно сохранна, за исключением перироландовой коры
• Таупатия также отмечается в глии: волокнистые астроциты, спиралевидные тельца в олигодендроцитах
д) Клиническая картина:
2. Демография:
• Возраст:
о Обычно 45-75 лет о Возрастной пик дебюта: 63 года
• Пол:
о Слабое преобладание у мужского пола
• Эпидемиология:
о 5-6 случаев на 100000
3. Течение и прогноз:
• Течение заболевания вариабельное
• Выживаемость с момента появления симптомов ПНП варьирует от 5,3 до 9,7 лет
• Нейропсихиатрические симптомы развиваются у > 50% пациентов в течении 2-х лет от начала заболевания
4. Лечение прогрессирующего надъядерного паралича:
• Симптоматическое
• Леводопа может оказать благоприятный терапевтический эффект при ригидности и брадикинезии при ПНП
• Терапия коферментом Q10 может оказать некоторый эффект

Болезнь Крейтцфельдта-Якоба – редкое дегенеративное заболевание центральной нервной системы, при котором происходит поражение коры и базальных ганглиев головного мозга, а также нарушение функций спинного мозга.
Болезнь Крейтцфельдта-Якоба (она же - губчатая энцефалопатия) встречается довольно редко и может длительное время протекать бессимптомно. Длительность инкубационного периода зависит от способа заражения: так, с момента заболевания до появления первых симптомов может пройти от 12 месяцев до 10-15 лет. Источником болезни, по распространенному мнению, считается коровье мясо, зараженное прионами*. Однако болезнь может носить наследственный характер или может быть занесена во время операции нестерильными медицинскими инструментами.
Особенности развития болезни Крейтцфельдта-Якоба
Как правило, болезнь включает три стадии развитии: продромальный период, инициальный и развернутый. Первая стадия (продромальная) характеризуется быстрой утомляемостью пациента, расстройством сна, отсутствием аппетита и прочими признаками, которые могут быть приняты, например, за проявление депрессии. Через несколько недель или месяцев наступает второй этап (инициальный), когда пациент испытывает головокружения и головные боли, ощущает ухудшение зрения.

Третья стадия, развернутая, может привести к спастическому параличу и таким формам сокращения мышц, как миоклония, тремор, ригидность. На этом этапе у пациента происходит атрофия нейронов головного мозга. Кроме того, пациент постоянно чувствует напряженность в мышцах. При некоторых клинических формах болезнь на третьей стадии сопровождается дисфункцией мозжечка.
Для достоверной диагностики данного заболевания наиболее эффективным методом является прижизненная биопсия с забором вещества мозга. Однако, как правило, биопсия назначается как крайний метод диагностики. МРТ в данном случае позволяет осуществить дифференциальную диагностику, что поможет специалисту исключить большинство различных патологий.
Болезнь Крейтцфельдта-Якоба: МРТ-диагностика
Для диагностирования дегенеративного заболевания лечащий врач может назначить комплекс взаимоуточняющих методик: ЭЭГ и МРТ головного мозга.
Магнитно-резонансная томография позволяет получить качественные и контрастные изображения головного мозга пациента. Во время проведения МРТ выявляются участки повышенного сигнала, локализованные в подкорковых ганглиях и таламусах. Кроме того, магнитно-резонансная томография дает возможность успешно определять признаки атрофических изменений мозжечка, коры головного мозга и расширение желудочковой системы.


МРТ является абсолютно безвредным и безболезненным методом диагностики. МРТ является абсолютно безвредным и безболезненным методом диагностики. Высокое контрастное разрешение позволяет распознавать любую патологию структур головного мозга, оценить состояние белого и серого вещества, черепно-мозговых нервов, ствола мозга, образований задней черепной ямки.
Магнитно-резонансная диагностика часто назначается в целях дифференциальной диагностики заболеваний нервной системы. Как мы уже отметили, болезнь Крейтцфельдта-Якоба встречается довольно редко и трудно идентифицируется, поэтому при постановке такого диагноза необходимо исключить другие заболевания головного мозга. Ими могут быть лобно-височная и мультинфарктная деменция, энцефалит, хронический менингит, нормотензивная гидроцефалия, энцефалопатия и другие заболевания, некоторые из которых, имеющие схожую с болезнью Крейтцфельдта-Якоба клиническую картину, вызывают деменцию. У пациентов с расстройством интеллекта (слабоумием) МРТ также выявляет структурные изменения в головном мозге и его аномалии.
Таким образом, при диагностике болезни Крейтцфельдта-Якоба важным преимуществом МРТ (в сравнении с другими методами лучевой диагностики) является возможность исключить большинство различных патологий.
Болезнь Крейтцфельдта-Якоба — редко встречающееся дегенеративное заболевание головного мозга, связанное с накоплением в нейронах патологического белка приона. Клинически болезнь Крейтцфельдта-Якоба проявляется слабоумием, пирамидными и экстрапирамидными нарушениями, миоклониями, симптомами поражения мозжечка и нарушением зрения. Диагноз болезни Крейтцфельдта-Якоба основывается на совокупности клинических симптомов, данных ЭЭГ, анализа цереброспинальной жидкости, МРТ и ПЭТ головного мозга, а также морфологического исследования образца тканей мозга, полученного в результате биопсии или посмертно. Эффективное лечение болезни Крейтцфельдта-Якоба пока не найдено. Заболевание имеет 100% летальный исход.
МКБ-10
Общие сведения
Причины
Установлено, что болезнь Крейтцфельдта-Якоба имеет инфекционный характер. Заражение может произойти при пересадке зараженных прионами тканей, через нейрохирургический инструмент и препараты крови, при введении некоторых гормональных препаратов (человеческого гонадотропина для лечения бесплодия и соматотропина для терапии гипопитуитаризма). Болезнь Крейтцфельдта-Якоба новой формы может развиваться после употребления в пищу мяса заболевших животных (коровы) или носителей инфекции (овец и коз).
В результате ряда исследований стало известно, что болезнь Крейтцфельдта-Якоба связана с проникновением в организм инфекционного белка — приона. В норме в клетках головного мозга человека содержится здоровый прион, имеющий несколько другое строение. Инфекционный прион, попадая в организм человека не разрушается, а с током крови поступает в головной мозг и откладывается на поверхности нейронов. Его взаимодействие с нормальными прионами мозговой клетки приводит к тому, что они изменяют свою структуру, постепенно трансформируясь в патогенную, подобную инфекционному приону, форму. Патогенные прионы образуют бляшки и приводят к гибели нейрона.
Болезнь Крейтцфельдта-Якоба имеет достаточно длительный инкубационный период, связанный с временем, необходимым для проникновения инфекционных прионов в мозговую ткань и патогенной трансформации здоровых прионов. Длительность инкубационного периода напрямую зависит от способа заражения. При инфицировании тканей головного мозга зараженным хирургическим инструментом болезнь Крейтцфельдта-Якоба развивается через 15-20 месяцев. При инфицировании через имплантированные в околомозговые структуры ткани (например, твердую мозговую оболочку, роговицу глаза) инкубационный период может длиться до 5,5 лет. При внутримышечном введении инфицированных лекарственных препаратов (например, гонадотропина, соматотропина, содержащих бычий тромбин гемостатиков) болезнь Крейтцфельдта-Якоба начинает проявляться спустя 12,5 лет.
Отмечаются также наследственные формы болезни Крейтцфельдта-Якоба, связанные с генетическими нарушениями, приводящими к образованию патологических прионов.
Классификация
Практическая неврология классифицирует болезнь Крейтцфельдта-Якоба с учетом ее клинической формы. В соответствии с этим выделяют: подострую спонгиоформную энцефалопатию, отличающуюся быстрым течением и диффузным поражением мозговой коры; классическую (дискинетическую) форму, представляющую собой сочетание пирамидных и экстрапирамидных симптомов со слабоумием (деменцией); промежуточную форму болезни Крейтцфельдта — Якоба, характеризующуюся преобладанием мозжечковых и подкорковых расстройств; амиотрофическую форму, двигательные и речевые расстройства при которой напоминают клинику бокового амиотрофического склероза.
Симптомы болезни Крейтцфельдта-Якоба
В большинстве случаев болезнь Крейтцфельдта-Якоба характеризуется постепенным развитием, однако возможно подострое или острое начало. Примерно в 30% случаев болезнь Крейтцфельдта-Якоба начинается с продромальных симптомов: раздражительности, рассеянности, головных болей, нарушений сна, головокружения, ухудшения памяти, снижения зрения, безынициативности, снижения либидо, изменения поведенческих реакций. Возможно эпизодическое возникновение эйфории и/или беспричинного страха, отрывистые бредовые или галлюцинаторные переживания. Из неврологических нарушений в продромальном периоде наблюдаются: шаткость во время ходьбы, парестезии, расстройство высших функций коры головного мозга (алексия, акалькулия и пр.). Описаны несколько случаев, когда болезнь Крейтцфельдта-Якоба дебютировала с появления корковой слепоты.
В стадии развернутых клинических проявлений болезнь Крейтцфельдта-Якоба характеризуется прогрессирующим спастическим параличем (параплегией или гемиплегией), атаксией, эпилептическими припадками. Возникают экстрапирамидные нарушения: мышечная ригидность, атетоз, тремор. Практически у всех больных наблюдаются миоклонии — быстрые неритмичные сокращения отдельных мышц. Чаще всего отмечается миоклонус губы и века. Наблюдаются вторично генерализованные миоклонические приступы. Появляется и нарастает ярко выраженная деменция, сопровождающаяся нарушениями речи вплоть до ее полного распада. Новый вариант болезни Крейтцфельдта-Якоба отличается преобладанием психиатрической симптоматики и расстройств чувствительности. В 100% случаев он сопровождается мозжечковыми нарушениями, в то время как при спорадической болезни Крейтцфельдта-Якоба расстройства функции мозжечка наблюдаются лишь в 40% случаев.
В терминальной стадии болезнь Крейтцфельдта-Якоба характеризуется глубокой деменцией. Пациенты не контактны, находятся в состоянии прострации, утрачен контроль над функцией тазовых органов. Наблюдаются гиперкинезы, выраженные мышечные атрофии, нарушения глотания, пролежни. Возможна гипертермия и эпилептические приступы. Смерть наступает в коматозном состоянии на фоне децеребрационной ригидности и выраженной кахексии.
Диагностика
Клиническая диагностика заболевания основана на сочетании прогрессирующей в течение 2-х лет деменции, пирамидных и экстрапирамидных расстройств, миоклоний, мозжечковых расстройств и нарушений зрения. Для уточнения диагноза невролог назначает инструментальные методы обследования: электроэнцефалографию (ЭЭГ), ПЭТ и МРТ головного мозга, люмбальную пункцию. В сомнительных случаях для установления диагноза болезнь Крейтцфельдта-Якоба производят стереотаксическую биопсию головного мозга.
На ЭЭГ на фоне сниженной биоэлектрической активности у большинства больных наблюдаются периодические или псевдопериодические острые волны. Отмечается билатеральная, фокальная или генерализованная миоклоническая активность, которая в начальной стадии определяется у половины больных, а в терминальной стадии выявляется в 100% случаев спорадической болезни Крейтцфельдта-Якоба. Новый вариант заболевания часто протекает без существенных изменений ЭЭГ-паттерна.
Люмбальная пункция в обязательном порядке проводится пациентам с подозрением на болезнь Крейтцфельдта-Якоба. Она позволяет оценить давление ликвора и произвести исследование цереброспинальной жидкости. Отсутствие патологических изменений ликвора позволяет дифференцировать болезнь Крейтцфельдта-Якоба от многих других заболеваний ЦНС.
Наиболее достоверным методом диагностики является морфологическое исследование образцов мозговой ткани, которые могут быть получены прижизненно путем биопсии или при аутопсии после смерти пациента. Применение иммуноцитохимического метода позволяет обнаружить в исследуемом материале отложения патологического белка — приона.
Дифференциальная диагностика
Дифференциальную диагностику болезни Крейтцфельдта-Якоба необходимо проводить с лобно-височной деменцией, герпевирусным энцефалитом, болезнью Альцгеймера, мультиинфарктной деменцией (слабоумием, развивающимся после повторных ишемических и геморрагических инсультов), хроническим менингитом, арахноидитом, нормотензивной гидроцефалией, энцефалопатией Хашимото, сопровождающей некоторые случаи аутоиммунного тиреоидита, и др.
Лечение болезни Крейтцфельдта-Якоба
В современной медицине подходы к лечению болезни Крейтцфельдта-Якоба находятся в стадии активной разработки. Общепринятые противовирусные препараты, а также пассивная иммунизация и вакцинация людей и животных выявились неэффективными. Отмечено, что блокирующее действие на синтез патологических прионов в инфицированных нейронах оказывает Брефелдин А, а блокаторы кальциевых каналов продлевают жизнь инфицированных клеток. Обычно пациенты, имеющие болезнь Крейтцфельдта-Якоба, получают симптоматическое лечение. Оно направлено на купирование миоклонических приступов и экстрапирамидных нарушений, в связи с чем применяются антиэпилептические и противопаркинсонические лекарственные средства.
Прогноз болезни Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба является фатальным заболеванием. Продолжительность жизни большинства больных не превышает 1 год с момента начала клинических проявлений; а средняя длительность составляет 8 месяцев. Лишь 5-10% заболевших живут в течение 2 и более лет. Наследственная болезнь Крейтцфельдта-Якоба в среднем длится около 26 месяцев.

Заболевания
Болезнь Крейтцфельдта–Якоба
Это быстро прогрессирующее фатальное нейродегенеративное заболевание, ключевым патогенетическим звеном которого является гибель нейронов, индуцированная прионовыми белками. Заболевание приводит к быстро прогрессирующему слабоумию и смерти обычно в течение года от начала.
История
Заболевание было названо в честь Ханса Герхарда Кройцфельдта (1885-1964), немецкого невролога, который впервые описал это состояние в 1920 году, и Альфонса Марии Якоба (1884-1931), немецкого невролога, которая также описала это состояние в отдельном исследовании в 1921 году.
Эпидемиология
Описаны три основных типа болезни Крейтцфельдта-Якоба:
- Спорадический – составляет 85-90% случаев и разделена на множество подтипов в зависимости от мутации;
- Семейный – 10% случаев, вызвана мутацией PRPc;
- Ятрогенный;
Особенности клиники
К основным клиническим проявлениям заболевания относятся: быстро прогрессирующие когнитивные нарушения, миоклонус, дистония, акинетикоригидный синдром, спастичность, гиперрефлексия, атаксия, зрительные расстройства, на поздних этапах заболевания акинетический мутизм. Примерно в трети случаев отмечаются эпилептические припадки. Средняя продолжительность жизни при спорадической форме БКЯ составляет около 5 мес; более 90% пациентов умирают в течение 1 года из-за аспирационной пневмонии в состоянии акинетического мутизма. Используемые в настоящее время диагностические критерии БКЯ включают быстро прогрессирующую деменцию, экстрапирамидно/ пирамидные и зрительные расстройства, миоклонус, мозжечковую атаксию, а также характерные изменения ЭЭГ в виде комплексов высокоамплитудных 2–3-фазных острых волн и положительный маркер белок 14-3-3 в цереброспинальной жидкости (ЦСЖ). Сложности диагностики БКЯ связывают с редкостью заболевания, клиническим полиморфизмом и недостаточной информированностью врачей.
Читайте также: