
Гликозилирование туберкулезных т антигенов
Обновлено: 19.04.2024
Нервная система при врожденных нарушениях гликозилирования
Гликозилирование белков является необходимым этапом котрансляционного созревания растущей полипептидной цепи. Данный процесс необходим для жизнеспособности и нормального развития. Это касается большинства внеклеточных белков, таких как белки сыворотки (трансферрин, α1-антитрипсин, α1-антихимотрипсин, некоторые факторы свертывания, апо-липопротеин С-III), большая часть мембранных белков, таких как рецепторы специализированных клеток, и некоторые внутриклеточные белки, такие как лизосомальные ферменты. Врожденные нарушения гликозилирования были подразделены на нарушения N-и О-гликозилирования.
Нарушения, связанные с дефектами маннозо-а-О-гликозилирования сочетаются с синдромом Уокера-Варбурга и различными формами мышечных дистрофий, таких как болезнь мышц-глазмозга, врожденная дистрофия Фукуяма и сочетанные миопатии (описаны в других разделах) (Muntoni et al., 2004; Martin, 2005). Нарушения N-гликозилирования приводят к мультисистемным аномалиям, которые обычно включают поражение центральной нервной системы. Заболевания данной группы подразделяются на два типа. Тип I врожденных нарушений гликозилирования характеризуется дефектом элонгации гликана, в то время как тип II врожденных нарушений гликозилирования связан с дефектом путей процессинга (см. обзор в работах Leroy, 2006; Sparks, 2006).
а) Биохимические изменения и патогенез. Гликозилирование белков является очень сложным процессом, в котором участвует множество ферментов и транспортеров.
Формирование необходимых олигосахаридов, также называемых гликанами, происходит в эндоплазматической сети. Различные доноры сахаров активируются в качестве связанных с нуклеотидами сахаров и долихил-фосфатами моносахаридов на цитоплазматической стороне эндоплазматической сети. Синтез олигосахаридов инициируется прикреплением двух N-ацетил-глюкозамин-фосфатных единиц к липидному носителю (долихол-фосфату), необходимому для процесса элонгации олигосахаридов.
В дальнейшем ступенчатая элонгация происходит за счет прикрепления маннозных и глюкозных остатков. Каждый шаг катализируется специфическими трансферазами, которые кодируются соответствующими ALG генами. При последнем шаге олигозид переносится на образующийся белок с помощью олигосахарид-протеин-гликозилтрансферазы. Различные врожденные дефекты данного пути являются причиной врожденных нарушений гликозилирования I типа.
После переноса, опосредованного олигосахарид-протеин-гликозилтрансферазой, процесс продолжается в аппарате Гольджи в виде удавления глюкозных и маннозных остатков под действием специфических глюкозидаз и маннозидаз. На данном этапе процесс разделяется на два пути. Один путь заключается в лизосомальном фосфорилировании, а его дефекты являются причиной муколипидоза II типа (болезнь I клеток) и III типа (псевдо-Гурлер синдром). Второй путь обеспечивает специфическую ориентацию белков относительно участка их функционирования, например, внутри плазматической мембраны или для секреции в межклеточное пространство. Данный путь, также называемый укороченным, заключается в удалении маннозных остатков с помощью специфичной маннозидазы и прикреплении двух N-ацетил-глюкозамин-фосфатных единиц с помощью специфичных трансфераз.
В дальнейшем происходит прикрепление одной фукозы, двух сиаловых кислот и двух галактоз. Для данного пути созревания необходимы специфические транспортеры фукозы и сиаловых кислот и галактозилтрансфераза. Дефекты укороченного пути являются причиной врожденных нарушений гликозилирования II типа. Описано два других врожденных нарушения гликозилирования II типа, вызванные дефектами сохраненных олигомеров Гольджи. Сохраненный олигомер Гольджи представляет собой белковый комплекс, фиксированный на мембране Гольджи и участвующий в сохранении и функционировании аппарата Гольджи.
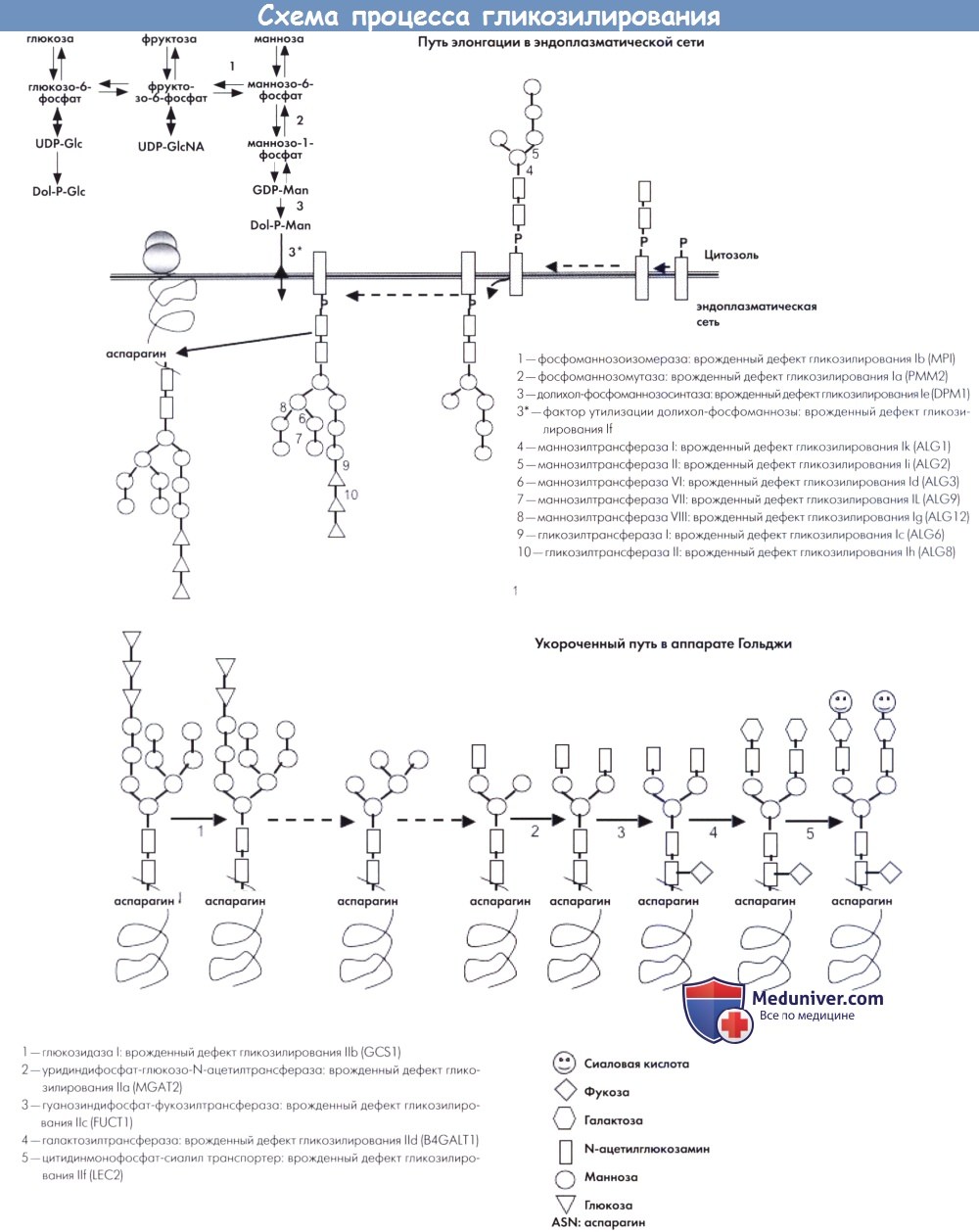
Схематическое описание процесса гликозилирования с указанием дефектов при врожденных нарушениях гликозилирования I и II типов:
(а) путь элонгации в эндоплазматической сети, (b) укороченный путь в аппарате Гольджи.
б) Диагностика. Диагностика врожденных нарушений гликозилирования проводится путем изоэлектрического фокусирования сывороточного трансферрина. Характеристики делятся на два типа. Тип 1 характеризуется повышением дисиало- и асиалотрансферриновых соединений одновременно со снижением тетрасиалотрансферрина. Тип 2 характеризуется увеличением трисиало- и моносиалотрансферриновых соединений. В дополнение к изофокусированию трансферрина изофокусирование других сиалиновых белков улучшает качество диагностики (Fang et al., 2004). Изоэлектрическое фокусирование аполипопротеина С-III используется для скрининга дефектов О-гликозилирования в отдельности или в сочетании с дефектами N-гликозилирования.
Выделяется три типа изоформ: аполипопротеин С-III0, аполипопротеин С-III1, аполипопротеин С-III2, в зависимости от количества остатков сиаловой кислоты, прикрепленных к гликопротеину (Wopereis et al., 2007).
в) Врожденные дефекты гликозилирования 1 типа. Врожденный дефект гликозилирования 1а типа (ВДГ-Iа) ВДГ-Ia является наиболее частым и наиболее изученным заболеванием из группы врожденных дефектов гликозилирования с частотой около 1 на 50000. Классические формы заболевания являются прогрессирующими и представлены тремя стадиями, развивающимися с возрастом (Hagberg et al., 1993; Stibler et al., 1994).
В младенческом периоде преобладают заметные мультисистемные поражения, которые прогрессируют от периода новорожденности до позднего младенческого возраста. Заболевание характеризуется плохим питанием, плохой прибавкой в весе и мышечной вялостью. Ранняя задержка психомоторного развития отмечается у всех пациентов в виде выраженной задержки развития крупной моторики, явной аксиальной гипотонии, мышечной слабости; позднее появляются признаки нарушения равновесия или истинной мозжечковой атаксии. Нормальная окружность головы при рождении может трансформироваться в приобретенную микроцефалию. Сухожильные рефлексы изначально слабые, арефлексия формируется в течение первых 2-3 лет жизни. Часто встречается зрительная невнимательность, блуждание взора и внутреннее косоглазие.
Врожденная катаракта встречалась в редких случаях. В большинстве случаев отмечался некоторый дисморфизм лица, втянутые соски и характерное строение тела с ограничением движений в суставах конечностей, деформацией грудной клетки и необычной липодистрофией со скоплением жира на ягодицах, в области промежности и/или на пальцах рук. Часто отмечаются липоатрофические полоски и бляшки. Слабо выраженная гепатомегалия сочетается с фиброзом печени или гигантоклеточным гепатитом. Отмечены случаи увеличения почек и протеинурии. Во многих случаях могут развиваться опасные эпизоды полиорганной недостаточности, которые включают тяжелые инфекции, печеночную недостаточность, выпот в перикард с тампонадой сердца и ступор, в отдельных случаях сочетающийся с припадками и внутричерепным кровоизлиянием. Данные острые эпизоды ухудшения являются причиной высокой детской смертности (15-20%).
В детском возрасте отмечается непрогрессирующая задержка умственного развития различной степени. У большинства пациентов коэффициенты DQ/IQ составляют около 50. Мозжечковая атаксия и периферическая нейропатия, способствующая мышечной атрофии, в большинстве случаев препятствуют самостоятельному передвижению. Возможны рецидивирующие эпизоды дискинетических или хореоатетозных движений. Обычно развивается косоглазие и пигментный ретинит. Инсультоподобные эпизоды, связанные с частичным тромбозом мозговых сосудов, развиваются у половины пациентов в возрасте после 4-5 лет и проявляются комой, припадками и транзиторной слепотой. Приблизительно у половины пациентов формируется эпилепсия.
В подростковом и взрослом возрасте состояние стабилизируется. Большинство пациентов овладевает рядом социальных навыков, но остаются иждивенцами.
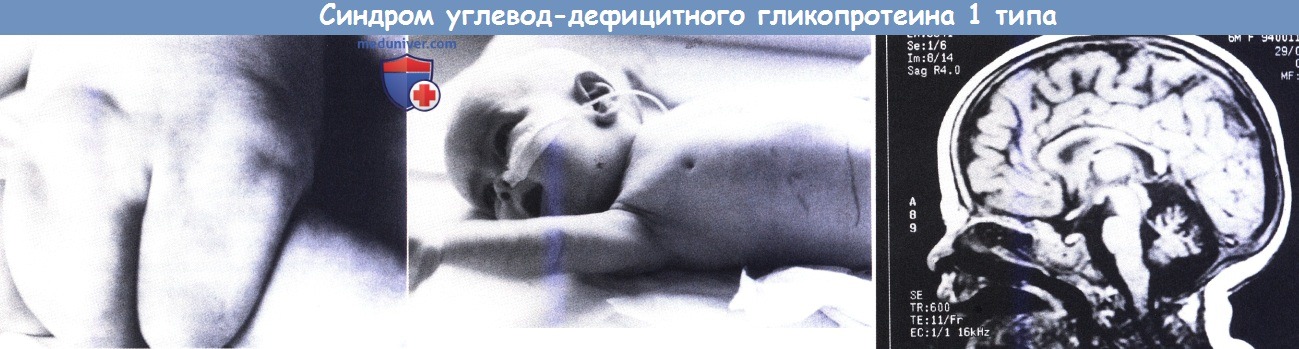
Синдром углевод-дефицитного гликопротеина 1 типа.
Слева: Аномальное отложение жира на ягодицах.
В центре: Втянутые соски; также отмечается затруднение питания (питание через зонд).
Справа: МРТ в сагиттальной проекции—заметная атрофия мозжечка и относительно тонкий ствол мозга.
Описанное течение заболевания касается большинства пациентов, тем не менее, описаны фенотипы с легкими или необычными проявлениями (Neumann et al., 2003; Coman et al., 2005; Noelle et al., 2005).
На ранних стадиях возможно умеренное повышение содержания белка в спинномозговой жидкости. Параметры электроэнцефалограммы неспецифичны. При электроретинографии отмечается прогрессирующее снижение ответа сетчатки (Andreasson et al., 1991). Зрительные, слуховые стволовые и соматосенсорные вызванные потенциалы могут отличаться от нормы. Скорость проводимости нерва снижена в моторных и сенсорных нервах, постепенное снижение отмечается вплоть до подросткового возраста.
При нейровизуализации у большинства пациентов в течение первого года жизни выявляется стремительно прогрессирующая гипоплазия мозжечка и ствола. Супратенториальные структуры обычно не отличаются от нормы, тем не менее, у трети пациентов отмечается центральная и/или корковая атрофия. Практически всегда отмечается оливопонтоцеребеллярная атрофия и миелиноподобные накопления в лизосомах (Eyskens et al., 1994). У двух близнецов, умерших в возрасте 4 месяцев и 6 лет, атрофия была более выражена при большей продолжительности жизни, что предполагает прогрессирование дегенеративного процесса. Очаговые инфаркты головного мозга предполагают окклюзию сосудов. При микроскопии выявляется полная утрата клеток Пуркинье, почти полная утрата гранулярных клеток в коре мозжечка и глиозядра моста (Stromme et al., 1991).
При биопсии икроножного нерва были выявлены аномальные слои миелина (Nordborg et al., 1991); отмечены лизосомальные накопления в клетках передних рогов (Eyskens et al., 1994), а также гепатоцитах, что свидетельствует об изменениях метаболизма миелина.
г) Другие врожденные нарушения гликозилирования 1 типа. Выявлено еще 11 вариантов нарушения путей элонгации олигосахаридов. Данным заболеваниям названия присвоены в алфавитном порядке (Ia—II) в соответствии с порядком описания. Аномалии развития и функции центральной нервной системы отмечаются при всех вариантах нарушений, за исключением одного (врожденное нарушение гликозилирования Ib). Врожденное нарушение гликозилирования-Ic является вторым по частоте заболеванием с клиническим фенотипом, сходным с врожденным нарушением гликози-лирования-Ia. Другие клинические фенотипы в настоящее время недостаточно описаны в связи с небольшим количеством пациентов в каждой группе. Некоторые характеризуются задержкой развития (от легкой до умеренной) с гипотонией, атаксией, косоглазием, нистагмом и припадками, варьирующими от фебрильных эпизодов до эпилепсии. У большинства пациентов отмечается тяжелая энцефалопатия новорожденных в сочетании с признаками, напоминающими врожденное нарушение гликозилирования-Iа.
У некоторых пациентов отмечается нейросенсорная тугоухость, слепота, колобома радужки и катаракта. Среди экстраневрологических симптомов часто встречается плохая прибавка веса, поражение скелета, кардиомиопатия или перикардиальный выпот, фиброз или цирроз печени и почечная недостаточность. В некоторых случаях выявляется водянка плода. Результаты МРТ могут быть нормальными или свидетельствовать о корковой и подкорковой атрофии, гипоплазии мозолистого тела или гипомиелинизации. Описаны случаи гипоплазии мозжечка, но данное проявление не является обязательным.
Врожденные нарушения гликозилирования 1 типа могут приводить к повреждению различных сывороточных гликопротеинов, оценка которых может способствовать диагностике. Низкое содержание свертывающих факторов и ингибиторов (фактора XI, антитромбина III, протеина С, протеина S, кофактора гепарина) может служить объяснением инсультоподобных эпизодов. В связи с неспецифическим просачиванием в сыворотку отмечается повышение содержание нескольких лизосомальных (например, аурилсульфатазы А, бета-гексаминидазы) и нелизосомальных ферментов (например, трансаминаз). Изменения также затрагивают многие транспортные белки (например, апопротеин В), гликопротеиновые гормоны (например, инсулиноподобный фактор роста-1) и факторы комплемента. Окончательный диагноз врожденного дефекта гликозилирования-1 подтверждается наличием сывороточных изоформ трансферрина 1 типа.
Учитывая данное обстоятельство, наиболее распространенные врожденные дефекты гликозилирования-Ia, Ib и Iс обычно диагностируются с помощью ферментных и молекулярных методов исследования. Другие типы заболевания диагностировались по результатам исследований, когда другие методы оказались неэффективны.
Врожденные нарушения гликозилирования I типа являются аутосомно-рецессивными заболеваниями, связанными с нарушением этапов сборки олигосахаридов. Все известные дефектные ферменты или транспортеры кодируются соответствующими генами; патогенетические мутации идентифицированы. Данная информация позволяет осуществлять достоверную пренатальную диагностику путем анализа мутаций в хорионической ДНК.
Эффективного лечения не существует.
д) Врожденные дефекты гликозилирования 2 типа. Врожденные дефекты гликозилирования 2 типа являются группой заболеваний, при которых затронут укороченный путь гликозилирования в аппарате Гольджи.
Врожденный дефект гликозилирования 2а типа Врожденный дефект гликозилирования 2а типа был первым описанным заболеванием данной группы. У небольшого количества пациентов отмечалась тяжелая задержка развития, отсутствие периферической нейропатии и нормальное строение мозжечка на МРТ. У пациентов отмечалась гипотония с младенческого возраста, генерализованные припадки с детского возраста и аномальное поведение со стереотипными движениями в виде потирания рук, давления языком и ударов головой. Все пациенты характеризовались низким ростом, скелетными деформациями, гипогонадизмом и небольшой печеночной недостаточностью. С биохимической точки зрения отмечается углеводный дефицит трансферрина со изоформой трансферрина 2 типа. Склонность к кровотечениям объясняется снижением содержания фактора свертывания XI. Недостаточность активности уридиндифосфат-глюкозо-N-ацетилтрансферазы II связана с мутациями гена MGAT2, расположенного на 14q21 хромосоме (Cormier-Daire et al., 2000).
Другие врожденные дефекты гликозилирования 2 типа Четыре других типа были связаны с дефектом действия глюкозидазы (IIb), галактозилтрансферазы (IId) и фукозы или транспортеров сиаловых кислот (IIc, IIf). Каждая группа заболеваний была представлена небольшим количеством или даже одним пациентом. Неврологические симптомы имели выраженный характер при IIb и IId типах заболевания и проявлялись в виде тяжелой энцефалопатии, некупируемых припадков и нормальной МРТ головного мозга (IIb) или умеренной задержки развития, мышечной слабости и мальформации Денди-Уокера (по результатам нейровизуализации) (IId). Тяжелые гематологические симптомы выявлялись при IIс и IIf типах заболевания. Результаты изоэлектрического фокусирования не отличались от нормы при всех типах заболевания, за исключением одного; врожденный дефект гликозилирования IIf типа сочетался с изменениями апопротеина-С-III1 (Wopereis et al., 2007).
Три других дефекта связаны с аномалиями комплекса сохраненного олигомера Гольджи (COG): CDG-IIe/COG7, CDG-IIg/COG1 и CDG-IIh/COG8. Результаты изоэлектрического фокусирования трансферрина плазмы 2 типа и свойств аполипопротеина-С-III0 свидетельствовали о повреждении N- и О-гликозилирования. Первое заболевание было описано у двух новорожденных сибсов, страдавших смертельной полиорганной недостаточностью и некупируемыми припадками. Дефект COG1 был выявлен у одного ребенка с дисморфическими проявлениями, некоторой задержкой развития и умеренной атрофией мозга и мозжечка при визуализации, выполненной в возрасте двух лет. У пациента с дефицитом COG8 отмечалась тяжелая энцефалопатия (Kranz et al., 2007).
Пероральное применение фукозы могло приводить к уменьшению выраженности гематологических изменений среди пациентов, страдающих врожденным дефектом гликозилирования Нс типа, но не оказывало воздействия на неврологический статус (Leroy, 2006). Для других типов заболевания специфического лечения не существует.
Ясно, что данная группа заболеваний недостаточно исследована и фенотипическая вариабельность среди уже описанных типов, а также дополнительные формы еще будут выделены после того, как станут известны все ферменты и соответствующие гены, задействованные в сложном процессинге гликозилирования.
ФГУ "НИИ пульмонологии" ФМБА России
ФГУ "НИИ пульмонологии" ФМБА России
Гистологическая дифференциальная диагностика гранулематозных болезней легких (часть II)
Журнал: Архив патологии. 2019;81(2): 59‑65
ФГУ "НИИ пульмонологии" ФМБА России
Гранулематозные болезни — это гетерогенная группа заболеваний различной этиологии, проявляющихся разнообразными клиническими синдромами и вариантами тканевых изменений, неоднородной чувствительностью к терапии и преобладанием общего доминирующего гистологического признака — наличия гранулем, определяющих клинико-морфологическую сущность каждой болезни. Гранулема является хронической воспалительной реакцией, в которой принимают участие клетки макрофагальной системы и другие клетки воспаления. После воздействия антигена происходит активация T-лимфоцитов, макрофагов, эпителиоидных клеток и гигантских многоядерных клеток, приводящая к образованию гранулемы. Гранулема включает также внеклеточный матрикс, продуцируемый фибробластами, позволяющий отграничить и изолировать антиген. Гранулематозные заболевания классифицируются по этиологии на инфекционные и неинфекционные. Однако, согласно последним исследованиям, патогенные микроорганизмы могут быть причиной развития гранулем при заболеваниях, ранее считавшихся неинфекционными. В ряде случаев классифицировать гранулематозный процесс как инфекционный и неинфекционный представляется затруднительным. Цель исследования — привлечь внимание читателей к разнообразию гранулематозных заболеваний, описать ключевые моменты патолого-анатомических проявлений различных болезней инфекционной природы, а также определить подход к дифференциальной диагностике гранулематозов.
ФГУ "НИИ пульмонологии" ФМБА России
ФГУ "НИИ пульмонологии" ФМБА России
Наиболее частой причиной развития гранулематозного процесса в легких является инфекция. Дифференциальная диагностика инфекционных гранулематозов основывается на характеристике гранулематозного воспаления и выявлении микроорганизма.
Грибы
Грибы, вызывающие глубокие микозы, как правило, не формируют гранулематозное воспаление в легких. В большинстве наблюдений такие грибы, как Aspergillus, Candida и некоторые другие, вызывают локальную мицетому, диффузный инвазивный микоз или аллергические реакции (аллергический бронхолегочный аспергиллез/микоз). Гранулематозные реакции, вызванные этими грибами, возникают редко [1].
Гистоплазмоз
Гистоплазмоз вызывается Histoplasma capsulatum (Северная Америка, долины рек) и H. duboisii (Африка), представляющими собой почкующиеся дрожжевые клетки диаметром 2—4 мкм. Микроорганизмы обнаруживают в цитоплазме макрофагов и гистиоцитов, а также и в некротическом детрите. Капсулы организма окрашиваются при окраске по Гимзе или ШИК-реакции. Формирование эпителиоидно-клеточных гранулем вызывают оба организма, однако некротизирующий гранулематоз чаще описан при H. сapsulatum [2].
Криптококкоз (европейский бластомикоз)

Cryptococcus neoformans встречается повсеместно, находится в почве, голубином помете, размер клетки 4—7 мкм, размножается почкованием, окрашивается гематоксилином и эозином, муцикармином, при ШИК-реакции. Криптококки вызывают различные изменения в легких. Типичная гранулематозная реакция представляет собой сливающиеся ненекротические гранулемы с множеством гигантских многоядерных клеток и нерезко выраженной воспалительной реакцией, гигантские клетки располагаются преимущественно вне гранулем и содержат клетки криптококков. Эти грибы можно также обнаружить внутри некротических гранулем (криптококкома), напоминающих таковые при микобактериальной инфекции и других видах грибов (рис. 1). Рис. 1. Фрагмент некротической гранулемы с округлыми образованиями диаметром 10—20 мкм (клетки криптококка). Окраска гематоксилином и эозином, ×400. У иммунокопрометированных лиц клетки криптококка можно обнаружить внутри альвеол, в их стенках и интерстиции при отсутствии выраженной воспалительной реакции, могут быть разрозненные гигантские многоядерные клетки [3].
Коккцидиоз
Coccidioides наиболее часто приводит к образованию некротической гранулемы, при этом эозинофильная реакция может быть выраженной или отсутствовать, нейтрофилы также могут быть многочисленны. Как и при других инфекциях, гранулемы располагаются перибронхиолярно или сообщаются с разрушенными бронхиолами. Процесс сопровождается формированием по периферии мелких ненекротизирующих гранулем. Coccidioides обычно находят в центре некротических гранулем, они состоят из больших сферических структур (сферул), содержащих дрожжеподобные структуры (эндоспоры), эндоспоры различного размера могут располагаться в некрозе или клеточном детрите, напоминая другие грибы. Наличие сферул и эндоспор свидетельствует в пользу коккцидиоза. Как и Histoplasma, Coccidioides не растут в культуре, таким образом, диагноз может быть установлен только при гистологическом исследовании [2].
Бластомикоз
Пневмоциста
Паразиты

К наиболее частым паразитам, способным приводить к развитию гранулематозного воспаления в легких, относится Dirofilaria. Этот круглый червь поражает чаще собак, но также встречается и у человека, заражение происходит путем укуса насекомыми. Личинка червя попадает в правые отделы сердца, при эмболии — в легочные артерии, вызывая тромбоз последних с развитием инфарктоподобных некрозов (рис. 3). Рис. 3. Личинка Dirofilaria в ветви легочной артерии, инфильтрация плазматическими клетками и эозинофилами. Окраска гематоксилином и эозином,×100. При этом в 1/3 наблюдений отмечается развитие гранулем в прилежащей ткани легкого, в половине — некротического или ненекротического васкулита, в 2/3 — эозинофильной инфильтрации [6].
Туберкулез

Туберкулезное воспаление вызывают члены семейства Mycobacterium tuberculosis, а именно M. tuberculosis, M. bovis, M. africanum, принадлежащие к группе быстрорастущих микобактерий. Вирулентность этих микобактерий варьирует от умеренных до высоковирулентных штаммов. В зависимости от вирулентности микобактерии, с одной стороны, и состояния иммунной защиты, с другой — изменения в легких при туберкулезе могут быть самыми разнообразными от распространенных некротических гранулем, милиарных некротических гранулем, ненекротических гранулем, туберкулемы, зажившей фиброзированной гранулемы (рис. 4) Рис. 4. Туберкулезная гранулема с лимфогистиоцитарным валом и гигантскими многоядерными клетками Пирогова—Лангханса по периферии. Окраска гематоксилином и эозином, ×100. [7]. Гранулемы при туберкулезе обычно бронхиолоцентричной локализации, но следует помнить, что они могут быть таковыми при любом инфекционном гранулематозе и даже при саркоидозе. Гистологические изменения при туберкулезе неотличимы от таковых при нетуберкулезном микобактериозе. Это было подтверждено в исследовании R. Corpe и I. Stergus [8], в котором 27 патологам, специалистам в диагностике микобактериальных заболеваний, было предложено оценить 25 гистологических препаратов без информации о культурально-подтвержденной инфекции. В большинстве случаев различить туберкулез или микобактериоз или не представлялось возможным, или сформулированный диагноз был ошибочным. Таким образом, диагностика туберкулеза должна быть основана на выявлении и последующем определении вида микобактерии! Сталкиваясь в процессе консультационной работы с пациентами, которые несколько месяцев (а иногда и не один год!) получали эмпирически назначенное лечение противотуберкулезными препаратами без подтверждения микобактериальной инфекции, полагаем, что такой подход приводит к повышению в нашей стране числа наблюдений с мультирезистентными штаммами микобактерий или штаммами с множественной лекарственной устойчивостью.
Нетуберкулезный микобактериоз
Нетуберкулезный микобактериоз — воспаление, вызываемое микобактериями, не относящимися к семейству микобактерий туберкулеза M. avium, M. fortuitum, M. gordonae, M. kansasii, M. xenopi и M. marinum, определяемых так же, как комплекс MAC. В отличие от микобактерий туберкулеза эти микобактерии могут быть обнаружены внутриклеточно в макрофагах (гистиоцитах), у иммунокопрометированных лиц они могут быть многочисленны. Диагностируются при проведении окраски на кислотоустойчивость, культуральных или молекулярно-биологических исследований. Как уже сказано выше, зачастую гистологические изменения сходны с таковыми при туберкулезе. Могут быть также выявлены ненекротические гранулемы, гистиоцитарные гранулемы, гранулемы, состоящие из пенистых, зернистых макрофагов, содержащих микобактерии. И.П. Соловьева и соавт. [9] описывают следующий спектр гистологических изменений при микобактериозах: туберкулезная гранулема — эпителиоидно-клеточная, число клеток Лангханса и интенсивность некроза варьируют, микобактерий немного; ареактивная, некротическая мультибациллярность — воспалительный ответ представлен слабо, в зоне некроза обилие микобактерий; мультибациллярный гистиоцитоз — диффузная макрофагальная инфильтрация с внутриклеточным обилием микобактерий, некрозы отсутствуют; мультибациллярный минимальный гистиоцитоз — слабая воспалительная реакция с внутриклеточным обилием микобактерий; гистиоидное поражение — узелковые скопления веретенообразных макрофагов с обилием микобактерий; неспецифическая грануляционная ткань; острый гнойный абсцесс.
Разнообразие заболеваний, приводящих к развитию гранулематозного воспаления, определяет определенные трудности в проведении дифференциальной диагностики даже при выполнении резекционных (операционных, видеоассистированных) биопсий, позволяющих получить достаточное количество материала для гистологического исследования [11]. Тем не менее установить причину гранулематозного воспаления удается не всегда. По данным T. Ulbright, A.-L. Katzenstein [12], проанализировавших 86 одиночных гранулем легких, выявленных при рентгенологическом исследовании, инфекционный процесс, вызванный кислотоустойчивыми микобактериями или грибами, был подтвержден в 70% случаев. В 25 наблюдениях инфекционная этиология не была доказана, при этом в 2 диагностирована гиалинизированная гранулема, в 1 — полиангиит с гранулематозом и в 22 классифицировать процесс не удалось. При этом было обнаружено значительное сходство гистологических изменений при инфекционных гранулемах и полиангиите с гранулематозом, вполне возможно, что последний явился отражением нарушений иммунного ответа на инфекционный агент, который уже не мог быть обнаружен в ткани. Это означает, что диагностировать полиангиит с гранулематозом и другие ангииты легких в случае солитарных узлов при отсутствии поражения других органов следует с крайней осторожностью. В таких случаях следует рекомендовать проведение тщательного обследования и динамического наблюдения за пациентами.
Данное исследование, на наш взгляд, представляется крайне интересным, поскольку прежде всего свидетельствует о преобладании саркоидоза и инфекционного гранулематозного воспаления в структуре гранулематозных заболеваний по данным гистологических исследований, проведенных в разных странах и географических регионах. Грибковая инфекция чаще явилась причиной гранулематозного воспаления в США, тогда как в других странах чаще была диагностирована микобактериальная инфекция, что является отражением эндемичности этих инфекций. Для улучшения качества этиологической диагностики следует обязательно направлять материал одновременно в гистологическую и микробиологическую лабораторию во всех наблюдениях, в которых подозревается гранулематозное заболевание. Причина гранулематозного воспаления, по данным этого исследования, не установлена более чем в трети наблюдений даже после гистологического исследования [10].

Проанализировав результаты консультативно-диагностических исследований нашей лаборатории, выявили, что число наблюдений, представляющих гранулематозные или гранулематозно-некротические процессы, составило практически треть от общего числа биопсий (284 на 1000 наблюдений) (см. табл. 2), Таблица 2. Частота встречаемости специфических гранулематозных инфекций [10] Примечание. *— микобактерия, выявленная при окраске на кислотоустойчивость в срезах, при отрицательном или невыполненном культуральном исследовании. в 36,2% наблюдений нам не удалось установить причину гранулематозного процесса в легких.
Частота инфекционного гранулематоза велика, исключая другие процессы, в диагностике которых существенную помощь оказывают данные анамнеза, клинических проявлений, лабораторных исследований и специфических морфологических изменений, о которых было сказано выше, остальные гранулематозные заболевания с наибольшей вероятностью следует относить к инфекционным.

Важным вопросом при дифференциальной диагностике инфекционных гранулематозов является выявление в срезах инфекционного агента. Для этого необходимо и обязательно применять дополнительные окраски. При выявлении грибковой инфекции прежде всего следует внимательно оценивать срезы, окрашенные гематоксилином и эозином. Большинство грибов, такие как Cryptococcus, Blastomyces, Coccidioides и Aspergillus, можно увидеть при окраске гематоксилином и эозином чаще в зонах некроза, чем в окружающей ткани. При проведении дополнительных окрасок следует выбирать срезы, в которых присутствует некроз. Как правило, для диагностики грибковой инфекции наиболее часто используют окраску серебрением по Грокотту, ШИК-реакцию, предлагают также окраски альциановым синим (по Моури), основным коричневым (по методу Шубича) или комбинированную окраску — ШИК-реакция с обработкой альциановым синим с докраской гематоксилином (рис. 5) Рис. 5. Клетки криптококка в гигантских многоядерных клетках в гранулеме. Комбинированная окраска альциановым синим, реактивом Шиффа и гематоксилином, ×200. [1].

Для диагностики микобактериальной инфекции используют окраску по Цилю—Нильсену, однако микобактерии, как правило, немногочисленны, их поиск трудоемок, использование альтернативных окрасок аурамином или аурамином/родамином повышает чувствительность метода, однако требует использования флюоресцентного микроскопа (рис. 6, а, Рис. 6. Кислотоустойчивые микобактерии. а — окраска по Цилю—Нильсену; б — аурамином/родамином (флюоресценция), ×1000. б). Для повышения выявляемости микобактерий T. Ulbright, A. Katzenstein рекомендуют проводить окрашивание по крайней мере с двух блоков [12]. При диагностике сифилиса рекомендуют проводить серебрение по Гомори или по Вартину—Старри [3].

При подозрении на гранулематозное заболевание и проведении резекции ткани необходимо оставлять часть ткани нефиксированной для возможного проведения культурального исследования и, если доступно, использовать метод быстрой заморозки при –70 °С для последующего проведения ДНК- и РНК-диагностики. При дифференциальной диагностике гранулематозных заболеваний следует прежде всего определить, является или гранулема инфекционной, или имеют место признаки других заболеваний, в том числе гранулематоза Вегенера. Если диагноз неинфекционной этиологии исключен, необходимо выполнить специальную окраску для обнаружения микроорганизмов, при этом желательно окрашивать срезы как минимум с двух блоков, при этом убедившись, что в материале присутствуют очаги некроза. Если на первый взгляд выявить микроорганизмы не удалось, рекомендуется просмотреть срезы еще раз при большем увеличении, а также использовать для окраски дополнительный блок. При отрицательном результате и клинических данных, свидетельствующих в пользу туберкулеза или других инфекций, рекомендуется проведение ПЦР. Если опять получен отрицательный результат, следует рекомендовать дополнительные культуральные и серологические исследования, исключающие инфекционный процесс (рис. 7). Рис. 7. Алгоритм диагностики некротических гранулематозных заболеваний легких [10]. Тем не менее по результатам этого алгоритма у определенной части гранулематозных заболеваний этиология не выяснена.
Таким образом, дифференциальная диагностика гранулематозных заболеваний легких сложна для патологоанатомов с целью достижения успеха желательно не только соблюдать правила исследования материала, приведенного в предложенном выше алгоритме, необходимы также тесное взаимодействие врача-пульмонолога, хирурга, выполняющего биопсию, а также коллегиальный подход.
Авторы заявляют об отсутствии конфликта интересов.
Сведения об авторах

Обзор
Автор
Редакторы

Спонсором приза зрительских симпатий выступил медико-генетический центр Genotek.
Тем не менее диагностика — это еще не диагноз, и результат любого теста не является истиной в последней инстанции. Диагноз же по-прежнему ставят, исходя из нескольких составляющих:
- клинической картины;
- наличия контакта с туберкулезным больным;
- рентгенографии легких, флюорографии или компьютерной томографии;
- результатов диагностических тестов.
Однако большинство этих методов диагностируют туберкулез постфактум — только скрининговые тесты помогают выделить лиц с высоким риском развития заболевания или с только зарождающимся процессом. И уже более 100 лет основным методом массовой диагностики туберкулеза является туберкулиновая проба Манту 2 ТЕ (содержащая 2 туберкулиновые единицы). Тест спорный, со множеством ложноположительных результатов, но именно его используют во всем мире и отказываться пока не собираются.
Проба Манту
В мире используют 3 вида туберкулина (рис. 1): датский препарат PPD (purified protein derivative) RT 23, американский PPD-S и российский — PPD-L. Различаются они видами микобактерий, из которых были получены: при производстве датского и американского препаратов используют только M. tuberculosis, а при производстве российского — смесь из M. tuberculosis и M. bovis (микобактерии, вызывающей туберкулез у крупного рогатого скота; на основе этого штамма была разработана вакцина БЦЖ). Различие в составах туберкулина обусловливает разные границы положительного результата: 15 мм для детей, привитых БЦЖ, и не более 10 мм для непривитых детей до 5 лет у датского препарата, не более 10 мм у американского и 5 мм у российского [4].

Рисунок 1. Препараты туберкулина
Диагностику с помощью пробы Манту проводят следующим образом: небольшое количество туберкулина вводят под кожу чуть выше запястья и через 72 часа оценивают реакцию, которую считают положительной, если в месте введения появляется припухлость (папула) более 5 мм в диаметре. В зависимости от ее размера, различают степень реакции от отрицательной (0–1 мм) до резковыраженной, или гиперергической (17 мм и более у детей и подростков, 21 мм и более у взрослых) [5]. И тут начинаются трудности, потому что у привитых БЦЖ реакция Манту положительна [6]! Мало того, чем больше поствакцинальный рубец, тем выше чувствительность к туберкулину [7]. Поэтому у привитых БЦЖ оценивают не только диаметр папулы, но и размер поствакцинального рубчика (табл. 1).
| Срок, прошедший с момента вакцинации БЦЖ | Размер рубчика после БЦЖ | Привитый иммунитет (мм) | Неясная причина | Подозрение на инфицирование |
|---|---|---|---|---|
| 1 год | 6–10 мм | 5–15 мм | 16 мм | Более 17 мм |
| 2–5 мм | 5–11 мм | 12–15 мм | Более 16 мм | |
| 0 мм | 2–4 мм | 5–11 мм | Более 12 мм | |
| 2 года | Вне зависимости от размера | Уменьшение размера папулы или прежний ее размер | Увеличение размера на 2–5 мм от предыдущего положительного результата | Реакция изменяется на положительную или папула увеличивается более чем на 5 мм |
| 3–5 лет | Вне зависимости от размера | 5–8 мм либо уменьшение размера папулы | Увеличение размера на 2–5 мм за последний год или отсутствие тенденции к уменьшению | Изменение на положительную (5 мм) реакцию или увеличение папулы на 6 мм; 12 мм при впервые поставленной пробе; изменение предыдущего размера на 2–4 мм или размер в 12 мм |
| 6–7 лет | Вне зависимости от размера | 0–4 мм | 5 мм | 6 мм и более |
| 7–9 лет | Если в 7 лет ребенку была сделана ревакцинация БЦЖ, реакция Манту вновь становится положительной и нормы повторяются | 0–4 мм | 5 мм | 6 мм и более |
| Взрослые | Отрицательная реакция, покраснение любого диаметра; папула до 4 мм | Более 5 мм |
Безусловно, метод оценки результата довольно субъективный. Но главное, папулу надо еще правильно измерить, зафиксировав только размер выпуклой части и игнорируя покраснение вокруг нее (рис. 2).

Рисунок 2. Правильная и неправильная оценки диаметра папулы
Еще одна трудность с диагностическими возможностями пробы Манту связана с тем, что они зависят от нижней границы положительного результата: чем ниже граница, тем больше будет наблюдаться ложноположительных реакций. К примеру, в России пороговым значением является папула в 5 мм. Это приводит к гипердиагностике — большому количеству зафиксированных реакций, но в итоге к низкому проценту реально выявленных случаев инфицирования [4].
В общем, не удивительно, что в последнее время информативность туберкулиновой пробы постоянно ставится под сомнение. Мало того, она может вызывать не связанные с туберкулезом аллергические реакции, поэтому все больше здоровых, но склонных к аллергии детей вынуждены идти в тубдиспансер в надежде выяснить причину положительной реакции. И наконец, на результат пробы может влиять множество факторов: недавно перенесенные инфекции, хронические заболевания, прием медикаментов, изменение гормонального фона или иммунитет к нетуберкулезным микобактериям [7].
Если говорить о достоинствах пробы Манту, то кроме низкой себестоимости, ими будут возможность выявления туберкулеза на ранних стадиях развития (за исключением лиц с иммуносупрессией и маленьких детей до двух лет [7]) и более высокая диагностическая ценность у непривитых БЦЖ по сравнению с привитыми (от 50% и ниже против 65,4% [6], [9]).
Мифы о пробе Манту

Рисунок 3. Антигены ESAT-6 и CFP-10
Что нужно знать о кожных пробах?
Анализы крови на туберкулез: плюсы и минусы
- в ранние сроки инфицирования;
- при иммунодефицитных состояниях;
- при неправильном заборе крови или ее транспортировке;
- из-за ошибок во время расшифровки результатов.
Добавлю, что тесты IGRA требуют наличия качественного оборудования, специальных реактивов и квалифицированного персонала, поэтому себестоимость у них довольно высока.
Кроме того, по сравнению с кожными тестами существенным недостатком этих анализов является определение in vitro только образования гамма-интерферона или активности Т-клеток. Поэтому в странах с высокой заболеваемостью (а Россия, несомненно, в их числе с показателем более 50 больных на 100 000 населения [13]) у IGRA-анализов нет никаких дополнительных преимуществ [12]. Тем более что их способность диагностировать туберкулезный процесс колеблется в районе 42–90% для разных возрастных групп, к тому же они не маркируют латентное носительство [12]. Другими словами, для жителей нашей страны анализы крови на туберкулез в общем-то напрасная трата денег, хотя в странах с низкой заболеваемостью (США, Канада, Западная Европа) они более информативны и рекомендованы к замене туберкулиновых проб для привитых БЦЖ [14].
В мире живет более полумиллиарда латентных носителей M. tuberculosis. Причем, далеко не каждый из них в итоге заболевает, потому что размножение микобактерии сдерживает иммунная система. Например, из 100 инфицированных палочкой Коха детей, лишь у одного развивается активная форма туберкулеза, поэтому диагностические тесты направлены не только на выявление латентных носителей, но и на оценку риска развития заболевания [15].
Анализы крови vs кожные тесты: сравнение эффективности
При оценке эффективности тестов для диагностики туберкулеза чаще всего учитывают два параметра: чувствительность (sensitivity) и специфичность (specificity). Под чувствительностью понимают способность метода выявлять лиц с заболеванием или носителей с высоким риском развития туберкулеза. Под специфичностью — способность теста правильно идентифицировать людей, у которых нет туберкулеза (то есть этот параметр характеризует риск появления ошибочных ложноположительных результатов) [12].
Какой тест все-таки выбрать?
Как говорится, при всем богатстве выбора альтернатив немного. К сожалению, при выборе теста многие руководствуются не его диагностическими характеристиками, а безвредностью для здоровья, потому что фенол в составе кожных проб пугает многих. И существует даже движение против пробы Манту, группа поддержки которого периодически предлагает заменить ее анализами крови IGRA. Но, как было отмечено выше, у подобных страхов нет никаких оснований. В придачу, являясь продуктом жизнедеятельности организма, фенол не накапливается, а выводится вместе с мочой. Поэтому главными критериями все-таки являются чувствительность и специфичность тестов.
Сведения об иммунной системе, межклеточных взаимодействиях в организме, накопленные за последние десятилетия, позволяют по-новому подойти к разработке патогенеза туберкулеза. Формирование полирезистентных штаммов микобактерий туберкулеза как один из итогов полувековой истории противотуберкулезной терапии и заметное увеличение числа больных с иммунодефицитами различной природы создают новые проблемы перед фтизиатрами всего мира. В настоящее время имеется насущная необходимость изучения патогенеза туберкулеза как в преморбидном периоде, так и при его прогрессирующем течении, а также изучения морфологических изменений в пораженных органах. Без продолжения таких исследований трудно понять причины современного патоморфоза туберкулеза, повышения уровня заболеваемости, терапевтических неудач и смертельных исходов.
The data on immunity and intercellular decades, allow one to have a further insight into the pathogenesis of tuberculosis. Formation of multiresistant strains of Mycobacterium tuberculosis as one of the results of semicentennial history of antituberculous therapy and a noticeable rise in the number of patients with immunodeficiencies of different etiology make phthisiologists in the world to be faced with new problems. There is now a vital need for studying the pathogenesis of tuberculosis both in its premorbidity and progression and for examining morphological changes in the affected organs. If these studies are not under way, it is difficult to understand the causes of the present-day pathomorphism of tuberculosis and higher rates of morbidity, mortality, and therapeutical failures.
А.Г. Хоменко — Центральный НИИ туберкулеза РАМН, Москва
A.H. Khomenko — Central Research Institute of Tuberculosis, Russian Academy of Medical Sciences, Moscow
Т уберкулез чаще всего развивается в результате заражения микобактериями человеческого вида, выделяемыми больным человеком. В ряде районов, неблагополучных по туберкулезу крупного рогатого скота, происходит заражение от животных, заболевание вызывается M. Bovis. Наиболее частый путь заражения – аэрогенный, но возможен алиментарный и весьма редко контактный, через поврежденную кожу или слизистые оболочки. Определенную защитную роль при аэрогенном заражении играет система мукоцилиарного клиренса, позволяющая частично вывести попавшие в бронхи частицы пыли, капельки слизи, слюны, мокроты, содержащие микроорганизмы. При энтеральном заражении может иметь значение всасывающая функция кишечника.
После проникновения микобактерий в организм человека, не зараженного ранее туберкулезом, в качестве первой защитной реакции развивается фагоцитоз. Эффективность этой защитной реакции зависит от многих факторов: возраста, пола, различных индивидуальных факторов риска, наследственной резистентности или предрасположенности к туберкулезу. Течение туберкулезной инфекции определяет в основном состояние иммунитета, как врожденного, так и приобретенного.
Процесс взаимодействия легочных макрофагов с микобактериями туберкулеза очень сложен и до конца не изучен. Результат этого взаимодействия определяется рядом механизмов, в том числе определяющих переваривающую способность макрофагов.
При недостаточной переваривающей способности макрофагов микобактерии туберкулеза могут сохраняться в них и даже размножаться, приводя к разрушению макрофагов и выходу из них микобактерий.
Макрофаги, фагоцитировавшие микобактерии, выделяют во внеклеточное пространство фрагменты разрушенных микобактерий, протеолитические ферменты, а также медиаторы (в том числе интерлейкин-1), которые взаимодействуют с Т-лимфоцитами, в частности Т-хелперами. Происходит активация Т-хелперов и выделение лимфокинов, в том числе интерлейкина-2, g-интерферона и других лимфокинов. Макрофаги устремляются к месту нахождения микобактерий, так как подавляется фактор угнетения миграции, выделяющийся b-лимфоцитами, под влиянием фактора активации макрофагов (этот фактор отождествляют с интерлейкином-2) возрастает ферментативная активность макрофагов. Активированные макрофаги выделяют также кожно-реактивный фактор, который обусловливает воспалительную реакцию, повышение сосудистой проницаемости. С этим фактором связывают появление повышенной чувствительности замедленного типа (ПЧЗТ) и положительной туберкулиновой реакции [1]. Кроме Т-хелперов значительное влияние на состояние иммунитета оказывают Т-супрессоры и супрессорные моноциты, которые угнетают иммунный ответ. Таким образом, процесс фагоцитоза и лизиса микобактерий регулируется Т-лимфоцитами. Их количественные изменения и функциональная активность в настоящее время хорошо изучены у больных туберкулезом, в том числе на субпопуляционном уровне [2, 3]. Кроме того, в инфекционном процессе активное участие принимают вещества, освобождающиеся при разрушении микобактерий. Удалось доказать, что корд-фактор (фактор вирулентности) микобактерий туберкулеза, обусловливающий их рост на плотной питательной среде в виде кос, провоцирует острый воспалительный процесс, а сульфатиды повышают токсичность корд-фактора и, главное, подавляют процесс образования фаголизосом в макрофагах, что предохраняет от разрушения расположенные внутриклеточно микобактерии. При интенсивном размножении микобактерий в организме человека вследствие малоэффективного фагоцитоза выделяется большое число токсичных веществ, индуцируется ПЧЗТ, которая способствует выраженному экссудативному компоненту воспаления с развитием казеозного некроза. В процессе разжижения казеозных масс микобактерии получают возможность для бурного внеклеточного размножения. Увеличивается число Т-супрессоров, происходит угнетение ПЧЗТ, снижение количества Т-хелперов, что приводит к анергии, обусловливающей прогрессирование туберкулезного процесса.
При сравнительно небольшой бактериальной популяции в условиях более эффективного фагоцитоза отмечается другая тканевая реакция – образование туберкулезной гранулемы и формирование туберкулезных очагов. Поскольку величина бактериальной популяции, а также характер течения иммунных реакций на разных этапах туберкулезной инфекции меняются, морфологические проявления у заболевших туберкулезом характеризуются чрезвычайно большим разнообразием.
Клинико-морфологические проявления первичного заражения микобактериями туберкулеза принято называть первичным туберкулезом. В настоящее время хорошо известно, что первичный туберкулез может проявляться не только в виде первичного туберкулезного комплекса, как это было принято считать ранее, возможно развитие туберкулеза внутригрудных лимфатических узлов, плеврита, различных изменений в легких – туберкулем, очагов и др. Первичный туберкулез в результате первичного заражения развивается лишь у 7 – 10% инфицированных, а остальные переносят первичную туберкулезную инфекцию без клинических проявлений, заражение проявляется лишь виражем туберкулиновых реакций. Отсутствие клинико-морфологических проявлений первичной туберкулезной инфекции может объясняться высоким уровнем естественной резистентности к туберкулезу, а также быть следствием иммунитета, приобретенного в результате противотуберкулезной вакцинации БЦЖ.
Первичный туберкулез может протекать с развитием распространенных или множественных изменений или ограниченных воспалительных. Прогрессирующее течение первичного туберкулеза проявляется преимущественно в виде милиарного туберкулеза и менингита, а также в виде первичной казеозной пневмонии с образованием каверны в легком. Такое течение первичной туберкулезной инфекции наблюдается редко и характерно для невакцинированных детей. В настоящее время реже, чем в прежние годы, отмечается хроническое течение первичной туберкулезной инфекции, протекающей у некоторых больных с наличием разнообразных параспецифических проявлений, так называемых масок туберкулеза [4, 5].
Заживление первичного туберкулеза может завершаться с выраженными или малыми остаточными изменениями. В настоящее время, как правило, первичный туберкулез излечивается с небольшими остаточными изменениями. У таких лиц развивается приобретенный иммунитет. Сохранение в остаточных очагах персистирующих микобактерий не только поддерживает приобретенный иммунитет, но и одновременно таит в себе риск эндогенной реактивации туберкулезного процесса вследствие реверсии измененных форм возбудителя туберкулеза в бактериальную форму и размножения бактериальной популяции.
В основе реактивации лежит прогрессирующее размножение микобактерий, находившихся в персистирующем состоянии. Установлено, что реактивация туберкулеза и развитие различных клинических форм вторичного туберкулеза чаще наблюдаются у лиц с остаточными изменениями при наличии факторов, ослабляющих иммунитет.
Возможен и другой путь развития вторичного туберкулеза – экзогенный, связанный с новым, повторным заражением микобактериями туберкулеза (суперинфекцией). Для развития вторичного туберкулеза помимо массивной повторной суперинфекции необходима совокупность ряда факторов, снижающих иммунитет. Вторичный туберкулез характеризуется большим разнообразием клинических форм.
Особо выделяют формы туберкулеза, возникшие вследствие бактериемии при проникновении микобактерий туберкулеза в кровяное русло и развития специфического васкулита. Заболевание может развиваться в виде острого или подострого диссеминированного туберкулеза с поражением многих органов – легких, плевры, печени, селезенки, гортани и др.
Различные сочетания морфологических реакций туберкулезного воспаления создают предпосылки для чрезвычайно большого разнообразия изменений в пораженных органах, особенно при хроническом течении болезни со сменой периодов обострения и затихания процесса. К этому следует добавить, что из сформировавшихся зон поражения микобактерии могут распространяться током лимфы или крови в непораженные участки и различные органы человеческого организма. Исход болезни зависит от течения болезни (прогрессирующего или регрессирующего), эффективности лечения и обратимости изменений, сформировавшихся в течение болезни. Доказано, что в условиях голодания и даже при недостаточном питании, особенно при недостаточном содержании в рационе белков и витаминов, нередко возникает реактивация туберкулеза. К факторам, способствующим реактивации, относятся также различные заболевания: сахарный диабет, лимфогранулематоз, силикоз, язвенная болезнь желудка и двенадцатиперстной кишки, состояние после резекции желудка, хронические воспалительные заболевания легких, психические заболевания, протекающие с депрессивным синдромом, алкоголизм, наркомания, стрессовые ситуации, синдром приобретенного иммунодефицита (СПИД), длительный прием глюкокортикоидов, цитостатиков. В настоящее время течение и исходы туберкулеза следует рассматривать только в условиях проводящейся противотуберкулезной химиотерапии. В процессе химиотерапии уменьшается микобактериальная популяция и создаются более благоприятные условия для репаративных процессов.
Отмечается разное течение инволютивного туберкулезного процесса: регрессия с последующим заживлением, стабилизация туберкулезного процесса без клинического излечения с сохранением каверны, туберкулемы или других изменений; временное затихание воспалительного процесса с последующим возникновением обострения. При неэффективном лечении могут иметь место хронизация или прогрессирование заболевания.
Заживление туберкулезного процесса и последующее излечение зависят не только от уменьшения бактериальной популяции, но и от способности микроорганизма обеспечить регрессию туберкулезного процесса.
При развитии хронического туберкулезного процесса с образованием каверн и появлением лекарственной резистентности микобактерий наступает прогрессирование заболевания, так как химиотерапия малоэффективна, подавления бактериальной популяции не происходит, наоборот, количество микобактерий увеличивается, поражаются все новые участки легких и другие органы. Такой прогрессирующий туберкулезный процесс может быть причиной смертельного исхода, особенно если у больного развивается казеозная пневмония или генерализованный туберкулез с поражением различных внутренних органов. Смертельные исходы заболевания туберкулезом обусловлены не только прогрессирующим течением хронических форм, но и нередким развитием в настоящее время изначально остро прогрессирующих форм туберкулеза, в прошлом названных скоротечной чахоткой. У взрослых это, как правило, казеозная пневмония с быстрым образованием множественных или гигантских каверн. Более чем у половины больных эта форма туберкулеза вызвана полирезистентными к противотуберкулезным препаратам микобактериями, а также осложнено неспецифической микрофлорой, повторяющимися легочными кровотечениями, дыхательной недостаточностью и выраженной интоксикацией.
Развитие остро прогрессирующих форм туберкулеза связывают с быстрым размножением микобактерий и образованием огромной бактериальной популяции, уничтожение или уменьшение которой с помощью противотуберкулезных препаратов невозможно вследствие имеющейся к ним полирезистентности микобактерий. Эта точка зрения подтверждается данными о внутригоспитальных эндемических вспышках туберкулеза со смертельными исходами в больницах Нью-Йорка в США. Следует также обратить внимание на подавление функционального состояния иммунокомпетентных систем организма, в частности моноцитарно-макрофагальной, которые в условиях иммунодефицитного состояния не могут осуществить защитную функцию. Роль иммунодефицита в развитии прогрессирующего туберкулеза и его смертельного исхода четко прослеживается у ВИЧ-инфицированных больных. Смертельные исходы наблюдаются также при сочетании прогрессирующего туберкулеза и рака, лейкоза, лимфогранулематоза.
Таким образом, если в недалеком прошлом изучение патогенеза туберкулеза проводилось в основном с точки зрения развития заболевания и его саногенеза, т.е. процессов заживления, в настоящее время имеется насущная необходимость изучения патогенеза туберкулеза как в преморбидном периоде, так и при его прогрессирующем течении, а также изучения морфологических изменений в пораженных органах. Без продолжения таких исследований трудно понять причины современного патоморфоза туберкулеза, повышения уровня заболеваемости, терапевтических неудач и смертельных исходов.
1. Медуницын Н.В., Литвинов В. И., Мороз А.М. Медиаторы клеточного иммунитета и межклеточного взаимодействия. – М., 1980.
2. Авербах М.М., Гергерт В.Я., Литвинов В.И. Повышенная чувствительность замедленного типа и инфекционный процесс. – М.,1974.
3. Туберкулез. Руководство для врачей // Под ред. А.Г.Хоменко. – М., 1996.
4. Иванова М.Г., Хмельницкий Б.Я. // Руководство по туберкулезу. – М., 1959. – Т. 2. – С. 241–77.
5. Струков А.И. Формы легочного туберкулеза в морфологическом освещении. – М., 1948.
Читайте также: