
Инфекционная молекула белка это
Обновлено: 24.04.2024
Для цитирования: Зуев В.А. Прионы – возбудители медленных инфекций человека и животных. РМЖ. 2010;6:381.
Схема процесса накопления молекул
инфекционного прионного
белка PrPSc
Литература
1. Sigurdsson B. Brit. Vet. J. 1954; 110: 255–270.
2. Idem Ibid: 307–322/
3. Ibem Ibid: 341–354/
4. Gajdusek D.C., Zigas V. New Engl. J. Med. 1957; 257: 974–978.
5. Gajdusek D.C., Gibbs C.J. Nature 1966; 209: 794–796.
6. Hadlow W.J. Lancet 1959; 2: 289–290.
7. Sigurdsson B., Thormar H., Palsson P.A. Arch. Ges. Virusforsch. 1960; 10: 368–381.
8. Horta–Barbosa L., Hamilton R., Writting B. et al. Nature 1969; 221: 974.
9. Зуев В.А. Медленные вирусные инфекции человеки животных.М., 1988.
10. Зуев В.А., Завалишин И. А., Ройхель В.М. Прионные болезни человека и животных (руководство для врачей). М., 1999.
11. Gajdusek D.C. Virology (Ed. B.N. Fields) New York, 1985; 1519–1557.
12. Gajdusek D.C. Subviral Pathogenesis of Plants and Animals:Viroids and Prions. New York 1985; 483–544.
13. Prusiner S.B., McKinley M.P., Groth D.F. et al. Proc. Natl. Acad. Sci. USA 1981; 78: 6675–6679.
14. Prusiner S.B. Science 1991; 252: 1515–1522.
15. Weissmann C. Nature 1991; 352: 679–683.
16. Bolton D.C., McKinley M.P., Prusiner S.B. Science 1982; 218: 1309–1311.
17. McKinley M.P., Bolton D.C., Prusiner S.B. Cell 1983; 35: 57–62.
18. Tobler J., Gaus T., Deboer P. et al. Nature 1996; 380: 639–642.
19. Harris D.A., Falls D.L., Johnson F.A. et al. Proc. Natl. Acad. Sci. USA 1991; 88: 7664–7668.
20. Oesch B., Westaway D., Walchli M., et al. Cell 1985; 40: 735–746.
21. Prusiner S.B. Neurodegenerative Diseases (eds. G. Golls & J.M. Stutzmann) Acad. Press 1996; 23–80.
22. Prusiner S.B. Prions Prions Prions (ed. S.B. Prusiner) Berlin 1998; 1–18.
23. Huang Z., Prusiner S.B., Cohen F.E. Prions Prions Prions (ed. S.B. Prusiner) Berlin 1998; 49–63.
24. Palmer M., Collinge J., Prion Diseases (eds. J. Collinge& M. Palmer) Oxford 1997; 1–17.
25. Collinge J., Palmer M. Prion Diseases (eds. J. Collinge & M.S. Palmer) Oxford 1997; 18–55.
26. Brown H. Antiviral Chem. Chemother. 1990; 1: 75–83.
27. Покровский В.И., Киселев О.И., Черкасский Б.Л. Прионы и прионные болезни. М., 2004.
28. Bruce M.E., Will R.G., Ironside J.M. et al. Nature 1997; 389: 498–501.
29. Hill A.F., Desbruslais M., Joiner S. et al. Ibid.: 448–450.
30. Григорьев В.Б., Покидышев А.Н., Кальнов С.Л., Клименко С.М. Вопр. вирусол. 2009; 5: 4–9.
31. Carrell R.W., Lomas D.A. Lancet 1997; 350: 134–138

Новость
Один из интегральных белков мембраны нейронов — предшественник токсичного пептида, вызывающего болезнь Альцгеймера, — играет определённую роль в росте и дифференциации нервных клеток. Однако фрагмент Aβ этого белка (β-амилоидный пептид, показан зелёным ), отщепляясь, приобретает способность к полимеризации и разрушает нервные клетки, что и приводит к возникновению болезни Альцгеймера.
Автор
Редакторы
Заболевания амилоидной природы — группа в основном неизлечимых прогрессирующих нейродегенеративных расстройств, включающая болезни Альцгеймера, Паркинсона и прионные заболевания. Молекулярный механизм этих болезней связывают со спонтанной пространственной перестройкой определённого белка (специфичного для каждого заболевания), придающей ему способность к полимеризации и образованию макромолекулярных фибрилл, токсичных для нервных клеток. Учёным из Чикаго удалось показать, что патогенным действием в болезни Альцгеймера обладают не только сами внутриклеточные фибриллы, но и предшествующие им сферические агрегаты, молекулы β-амилоидного пептида (Aβ) в которых имеют весьма схожую упаковку с фибрилло-образующими формами. β-структурные элементы, обнаруженные в этих молекулах, могут оказаться определяющим фактором для приобретения пептидом токсического действия.
Механизм болезни Альцгеймера, как и других нейродегенеративных заболеваних амилоидной природы, принято объяснять конформационной перестройки определённого белка, специфичного для конкретного заболевания. Белок болезни Альцгеймера (Aβ) состоит из примерно 40 аминокислотных остатков и является продуктом расщепления белка-предшественника, расположенного в мембране нервных клеток. Несмотря на существенные различия амилоидных белков из разных организмов и характерных для разных заболеваний, считается, что механизм всех этих болезней примерно одинаков (см. врезку). Многие амилоидные пептиды, безвредные в мономерной форме, становятся токсичными после полимеризации и образования фибрилл. Вследствие этого, нейротоксичность считается прямым следствием фибриллизации белка.
Прионы
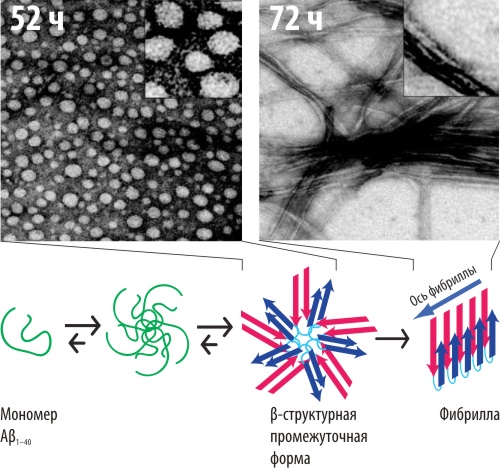
Рисунок 1. Промежуточные стадии формирования амилоидной фибриллы. Вверху: Морфологические свойства амилоидных образований, наблюдаемых через различное время инкубации мономерной формы Aβ (трансмиссионная электронная микрофотография). Промежуточные стадии характеризуются сферическими агломератами, конечная — образованием протяжённых фибрилл. Внизу: предложенный учёными механизм формирования фибриллы. Мономерная форма Aβ, не содержащая в своей структуре β-тяжей, ассоциирует в частицы сферической формы (~10 нм), в составе которых молекулы альцгеймеровского пептида меняют конформацию (возникающие β-структуры показаны стрелками), что приводит к укрупнению частиц до ~20 нм. Эти частицы (формирующиеся через 52 часа после начала инкубации) уже обладают токсическим действием и через некоторое время полимеризуются с образованием фибрилл.

Инфекционные болезни могут развиваться не только из-за бактерий, вирусов и других хорошо известных врачам микроорганизмов - это ошеломляющее заявление в медицинской среде было сделано после того, как в 1982 году профессор неврологии и биохимии Стэнли Прузинер (США) обнаружил белковые соединения, способные вызывать заболевания. Открытие белков-прионов было настоящим прорывом в медицине, доказательством чему стало получение учёным Нобелевской премии в 1997 году.
Прионы: биологическая сущность, свойства, среда обитания таинственных молекул
Как можно заразиться прионами?
На сегодняшний день выделяют следующие основные пути заражения инфекционным белком-прионом:
1. Трансмиссивный. В этом случае молекулы белка передаются от одного вида млекопитающего к другому - например, от инфицированной коровы или овцы человеку. Заражение происходит при употреблении в пищу мяса или молока заражённого животного, либо использовании его тканей (роговицы, препаратов крови и т.п.), применении во время оперативных вмешательств биологического шовного материала.
2. Наследственный. Заболевание развивается на фоне генетической мутации, затрагивающей область 20-й хромосомы. Несмотря на слабую изученность функционирования этого участка генома, достоверно известно его участие в синтезе нормального прионного белка. В случае генных мутаций вместо здорового приона образуется патологический, а это приводит к развитию болезней.
3. Спорадический. При этом аномальный белок появляется в организме спонтанно, без видимых причин.
Вне зависимости от способа появления аномальный белок может стать причиной заражения других людей.
Прионные заболевания: особенности течения, лечения, прогноз
Отличительной особенностью болезней, вызываемых прионами, является длительный инкубационный период - от 2-3 месяцев до нескольких десятилетий. Подавляющее большинство прионных заболеваний человека являются спорадическими и имеют семейный характер наследования.
Куру, синдром Герстманна-Штреусслера-Шейнкера, болезнь Крейтцфельдта-Якоба, скрэпи – прионы вызывают заболевания, сопровождаемые поражением центральной нервной системы. Для них характерны такие признаки как деменция (слабоумие), зрительные и мозжечковые нарушения. При этом у больного могут отмечаться двигательные расстройства, бессонница, галлюцинации, нарушение речи.
К сожалению, эффективных методов лечения прионных болезней на сегодняшний день нет, хотя учёные пытаются предотвращать переход нормального белка в аномальный. Пациентам назначается симптоматическая терапия с использованием противосудорожных средств для облегчения страданий. Прогноз пока неутешителен, так как все вышеперечисленные заболевания завершаются летальным исходом.
Перспективы
Раскрытие загадочных явлений, которыми окутаны прионы, возможно, поможет в понимании ряда серьёзных биомедицинских проблем человечества.

Новость
Путь прионов
Автор
Редакторы

Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Биологическая сущность прионов
Рисунок 1. Метафора нейродегенеративного поражения мозга — это губка, в которую превращается нервная ткань в результате массовой гибели нейронов.
И тогда происходит удивительное событие: нормальные молекулы белка, контактируя с патологическими, сами превращаются в них, изменяя свою пространственную структуру (механизм трансформации остаётся загадкой и по сей день) [1]. Таким образом прион, как самый настоящий инфекционный агент, заражает нормальные молекулы, запуская цепную реакцию, разрушительную для клетки.
Некоторые сведения о прионах
Условия возникновения заболеваний
Условия возникновения прионовых болезней уникальны. Они могут формироваться по трём сценариям: как инфекционные, спорадические и наследственные поражения. В последнем варианте главную роль играет генетическая предрасположенность [2].
В последнее десятилетие интерес к этой теме возобновился в связи с возможностью развития диагностики и эффективной терапии [5]. Появилось множество различных объяснений для возрастных нейродегенеративных болезней, — например, окислительная модификация ДНК, липидов и/или белков; соматические мутации; измененный врождённый иммунитет; экзогенные токсины; несоответствия ДНК—РНК; нарушение работы шаперонов; отсутствие одного из аллелей гена [5]. Альтернативным комплексным разъяснением служит то, что различные группы белков могут формировать прионы. Несмотря на то, что небольшое количество прионов может быть удалено посредством путей белковой деградации, их чрезмерное накопление с течением времени позволяет прионам самостоятельно распространяться в организме (рис. 2), что приводит к нарушению деятельности центральной нервной системы [5].

Группы риска прионных заболеваний
Вот кого прионные заболевания могут настичь с наибольшей вероятностью:
- работники пищевой промышленности;
- ветеринары;
- патологоанатомы;
- хирурги;
- пациенты трансплантолога;
- каннибалы;
- лица, в семье которых были замечены синдромы Герстманна—Штрейслера—Шейнклера или фатальной инсомнии.
Лабораторная диагностика и лечение
Диагностика базируется на внутримозговом заражении мышат или хомяков, у которых медленно (до 150 дней) развивается соответствующее заболевание, если пациент был болен [2]. Часто проводится гистологическое исследование головного мозга погибших животных [2].
К сожалению, до настоящего времени еще не разработаны эффективные методы лечения прионовых болезней, хотя попытки предотвратить конформационный переход нормального белка в аномальный производятся. Поэтому самым надёжным способом предупреждения развития инфекционных форм является профилактика [2].
Перспективы
По-видимому, интерес к прионам не угаснет до тех пор, пока предположения на их счёт полностью не подтвердятся и не будут найдены эффективные способы лечения прионных заболеваний. В статье [6] говорится о необходимости современного исследования, которое требует тщательного рассмотрения чужеродных прионов в экстраневрональных тканях.
В качестве модельных объектов авторы использовали мышей: две линии, которые трансгенно экспрессировали овечий прионный белок, и одну линию, которая экспрессировала человеческий прионный белок (рис. 3). Задачей было сравнить эффективность межвидовой передачи инфекции посредством тканей мозга и селезёнки. Внутримозговое заражение чужеродным прионным белком выражалось в отсутствии или небольшом количестве инфекционного агента в мозгах этих мышей. Однако инфекционные чужеродные прионы обнаруживались в селезёнке на более ранних этапах заражения в сравнении с моментом, когда были использованы нейротропные прионы, тем самым определяя, что лимфатическая ткань может быть более пермиссивной к распространению чужеродных прионов по сравнению с мозгом.

Рисунок 3. Способность приона хомяков Sc237 заражать и передаваться при введении в мозг или селезенку трансгенным мышам, имеющим прионный белок PrP овцы (tg338; белые мыши) или человека (tg7; серые мыши). Число заболевших/инъецированных мышей показано в скобках; ниже приведено среднее время жизни (в днях).
Чем вызвана эта предпочтительная репликация прионов в лимфатических тканях, пока неизвестно. Однако полученные данные показывают, что человек может быть более чувствительным к чужеродным прионам, чем предполагалось ранее на основании присутствия прионов в мозгу, и по этой причине бессимптомный переносчик прионной болезни может быть не распознан. Это ещё раз подтверждает, что такая могущественная биомолекула как прион таит в себе немало загадок, раскрытие которых, возможно, поможет в понимании ряда неразрешимых проблем человечества.
История

Гистологический препарат головного мозга, на котором видны микрополости


Как уже было сказано выше, основы знаний о прионах заложил Стенли Прузинер. Немного из его биографии. Родился в США в 1942 году. Его предки - эмигранты из российской империи, еврейского происхождения, вынужденные покинуть страну из-за еврейских погромов. Сам Стенли Прузинер в 1968 г. закончил Университет Пенсильвании и работал ординатором-неврологом в Медицинской школе Калифорнийского университета (Сан-Франциско). В 1970 впервые встретился с болезнью Крейтцфельдта — Якоба. У пациента, находившегося на лечении у Прузинера, никак не выявлялся возбудитель. Плотно занявшись этим исследованием, невролог обратился к трудам другого врача – Сиггурдсона, выявившего определенные закономерности у непонятных на тот момент болезней.
Такими закономерностями стали:
- необычно продолжительный (месяцы и годы) инкубационный период;
- медленно прогрессирующий характер течения;
- необычность поражения органов и тканей;
- неизбежность смертельного исхода.
Что же такое прионы и каков их механизм действия на организм (современные представления)?
На самом деле в организме человека и многих других живых существ есть белки PrPC. По-русски – нормальная форма прионных белков (открыты были после исследований Сиггурдсона, поэтому такая странность в название). Известна его длина, последовательность аминокислот, вторичная структура. Важно знать, что конечная структура состоит из трёх α-спиралей и двухцепочечного антипараллельного β-листа. Обладают интересным свойством, а именно осаждаются высокоскоростным центрифугированием, что является стандартным тестом на наличие прионов. Есть данные, что PrP играет важную роль в прикреплении клеток, передаче внутриклеточных сигналов, а потому может быть вовлечён в коммуникацию клеток мозга. Тем не менее, функции PrP исследованы недостаточно.

(a) норма (b) патология
Считается, что прионное заболевание может быть приобретено 3 путями: в случае прямого заражения, наследственно или спорадически (спонтанно) или их комбинациями. Спорадическая (то есть спонтанная) прионная болезнь возникает в популяции у случайной особи. Таков, например, классический вариант болезни Крейтцфельдта — Якоба. Существуют две основные гипотезы относительно спонтанного появления прионных болезней. Согласно первой из них спонтанное изменение происходит в самом доселе нормальном белке в мозге, то есть имеет место посттрансляционная модификация. Альтернативная гипотеза гласит, что одна или несколько клеток организма в какой-то момент претерпевают соматическую мутацию (то есть, не передающуюся наследственно) и начинают производить дефектный белок PrPSc. Как бы то ни было, конкретный механизм спонтанного возникновения прионных болезней неизвестен. Вторая – заражение. По данным современных исследований, основной путь приобретения прионных заболеваний — поедание заражённой пищи. Считается, что прионы могут оставаться в окружающей среде в останках мёртвых животных, а также присутствуют в моче, слюне и других жидкостях и тканях тела (кровь, ликвор). Из-за этого заражение прионами может произойти и в ходе пользования нестерильными хирургическими инструментами. Это усложняет стерилизацию хирургических инструментов или устройств на скотобойне. Прионы в большинстве своём устойчивы к протеазам, высокой температуре, радиации и хранению в формалине, хотя эти меры и снижают их способность к заражению. Эффективная дезинфекция против прионов должна включать гидролиз или повреждение/разрушение их третичной структуры. Это можно достичь обработкой хлорной известью, гидроксидом натрия и сильнокислыми моющими веществами. Пребывание в течение 18 минут при температуре 134 °C в герметичном паровом автоклаве не может деактивировать прионы. В качестве основного современного метода для деактивации и денатурации прионов в настоящее время изучается озоновая стерилизация. Ренатурация полностью денатурированного приона до инфективного состояния зафиксирована не была, однако для частично денатурированных прионов в некоторых искусственных условиях это возможно. Еще стоит помнить, что эти белки могут долго сохраняться в почве за счёт связывания с глиной и другими почвенными минералами. Не впадайте в паранойю, но теоретически они могут быть повсюду. В 2011 году было сообщено об открытии прионов, передающихся по воздуху в частицах аэрозоля (то есть воздушно-капельным путём). Также в 2011 году было опубликовано предварительное доказательство того, что прионы могут передаваться с получаемым из мочи человеческим менопаузальным гонадотропином, применяемым для лечения бесплодия. Теоретически с помощью всего одного больного животного с прионной болезнью, можно уничтожать целые нации и страны, просто добавляя его костную муку в кормовые добавки и продавая их в нужное государство. Сходная ситуация произошла в конце 80-х годов в Британии (эпидемия коровьего бешенства). Тогда, скорее всего по незнанию (а не по злому умыслу) произошел вышеуказанный процесс, унесший жизни около 200 человек (на 2009 год) и 179 тыс. голов крупного рогатого скота.

Клиника
Поговорим о болезнях и клинических проявлениях. Теоретически может возникать у всех живых существ, обладающих PrPc Вот некоторые примеры. У овец и коз, как это уже говорилось выше, главное проявление - это скрейпи. Для коров характерно коровье бешенство (губчатая энцефалопатия крупного рогатого скота) У норок- Трансмиссивная энцефалопатия норок. И так далее. Зафиксированы проявления заболеваний у кошек, диких парнокопытных, страусов. Но нас интересуют болезни человека.
Болезнь Крейтцфельдта — Якоба. Код по МКБ-10 A81.0; F02.1. Код А соответствует инфекционным болезням (А81 – инфекционные болезни нервной системы). Код F – психические расстройства, F02 – деменции.

Темно зеленый распространение К-Я
Светло зеленый - коровьего бешенства
Основные клинические критерии для постановки диагноза:
Выделяют несколько клинических форм:
Спонтанная — классическая (sCJD) Согласно современным представлениям (прионной теории), прионы при этой форме заболевания возникают в мозге спонтанно, без какой-либо видимой внешней причины. Болезнь обычно поражает людей в возрасте старше 50 лет и проявляется с вероятностью 1-2 случая на миллион жителей. Вначале проявляется в форме кратких потерь памяти, изменениями настроения, потерей интереса к происходящему вокруг. Далее симптомы деменции прогрессируют со всеми вытекающими последствиями.
Наследственная (fCJD) Болезнь возникает в семьях, где наследуется повреждение гена для прионового белка. Дефектный прионовый белок является намного более подверженным спонтанному превращению в прион. Признаки и ход болезни подобны классической форме.
Ятрогенная (1CJD) Болезнь обусловлена непреднамеренным внесением прионов в тело пациента при медицинском вмешательстве. Источником прионов ранее были некоторые лекарства, инструменты или мозговые оболочки, которые забирались у мертвых людей и использовались для закрытия раны при операциях на мозге. Признаки и ход болезни подобны классической форме. Новый вариант (nvCJD) Болезнь появилась впервые в 1995 году в Великобритании и с того момента от нее умерло не более 100 человек. Вероятнее всего, что они заразились мясными продуктами, содержащими бычьи прионы.
- психические расстройства и сенсорные нарушения,
- характерны глобальные когнитивные нарушения и атаксия.
- описано несколько случаев заболевания, дебютировавшего с корковой слепоты (вариант Heidenhain).
- эписиндром представлен также миоклоническими припадками.
- мозжечковая симптоматика выявляется в 100 %.
- Пациент страдает от всё более тяжёлой бессонницы, панических атак и фобий. Эта стадия длится в среднем 4 месяца.
- Панические атаки становятся серьёзной проблемой, и к ним присоединяются галлюцинации. Эта стадия длится в среднем 5 месяцев.
- Полная неспособность спать, сопровождаемая быстрой потерей веса. Эта стадия длится в среднем 3 месяца.
- Пациент перестаёт говорить и не реагирует на окружающее. Это последняя стадия болезни, длящаяся в среднем 6 месяцев, после чего пациент умирает.
Куру, почти не встречается в настоящее время, в связи с искоренением каннибализма. Интересно, что в 2009 году американские учёные сделали неожиданное открытие: некоторые члены племени форе, благодаря появившемуся у них в сравнительно недавнем времени новому полиморфизму гена PRNP, имеют врождённый иммунитет к куру.
В настоящее время нет ни одного средства останавливающего или тормозящего развитие прионных болезней.
Читайте также: