
Клинические формы энцефалита шильдера
Обновлено: 18.04.2024
Лейкоэнцефалит Шильдера — дегенеративно-демиелинизирующее поражение головного мозга, сопровождающееся образованием крупных или сливных зон демиелинизации. Имеет неуклонно прогрессирующее течение с неспецифичной и полиморфной клинической картиной, которая может включать психические нарушения, пирамидный и экстрапирамидный синдромы, когнитивный дефицит, поражение черепно-мозговых нервов, эписиндром. Диагностируется лейкоэнцефалит Шильдера по клиническим критериям и результатам МРТ после исключения другой патологии с подобными проявлениями. Терапия осуществляется глюкокортикостероидами, антиконвульсантами, миорелаксантами и психотропными средствами. Однако лечение малоэффективно.
МКБ-10
Общие сведения
Преимущественный возраст манифестации болезни Шильдера до сих пор остается спорным вопросом. Зарубежные специалисты в области неврологии считают характерным дебют в возрастном периоде от 7 до 12 лет, а отдельные авторы предлагают относить заболевание к детской форме рассеянного склероза. Наблюдения отечественных неврологов, напротив, свидетельствуют о равной степени поражения лиц различной возрастной категории.
Причины лейкоэнцефалита Шильдера
Этиопатогенез болезни Шильдера находится в стадии изучения. Из названия заболевания видно, что первоначально подразумевалась воспалительная этиология церебрального поражения, т. е. энцефалит. Предполагается вирусная теория заболевания по типу медленных инфекций. Среди возможных инфекционных агентов дискутируется роль кори, герпетической инфекции, миксовирусов, которые, возможно, запускают процесс аутоиммунного церебрального воспаления. Однако безуспешные попытки выделения возбудителя привели к возникновению иной этиопатогенетической теории. Последняя предполагает связь лейкоэнцефалита Шильдера с дисфункцией регуляторных механизмов липидного обмена, что сближает заболевание с наследственными лейкодистрофиями.
Морфологические изменения заключаются в образовании в белом церебральном веществе полушарий значительных зон демиелинизации, имеющих четкие заостренные очертания и зачастую асимметрично расположенных. В ряде случаев подобные очаги формируются в мозжечке и мозговом стволе. У пациентов, заболевших в пубертатном периоде и во взрослом возрасте, описаны случаи, когда наряду с зонами обширной демиелинизации наблюдаются округлые бляшковидные очаги, напоминающие бляшки рассеянного склероза.
Симптомы лейкоэнцефалита Шильдера
Заболевание отличается наличием неспецифичной и полиморфной симптоматики. Может манифестировать исподволь развивающимися психическими расстройствами: лабильностью настроения, апатией, нарушением поведения, эпизодами возбуждения с галлюцинаторным синдромом. Интеллектуальное снижение прогрессирует вплоть до деменции. Наблюдаются аграфия, акалькулия, алексия, агнозия, апраксия. Вследствие демиелинизации черепных нервов возникает неврит зрительного нерва, офтальмоплегия, тугоухость, снижение зрения, бульбарные расстройства. При поражении мозжечка появляется мозжечковая атаксия, скандированная речь, интенционный тремор. Поражение зрительной зоны коры проводит к гемианопсии, корковому амаврозу. Возможны экстрапирамидные нарушения в виде гиперкинезов, торсионной дистонии и т. п. Пирамидные расстройства обычно наблюдаются на поздних этапах лейкоэнцефалита в виде моно-, геми- и тетрапарезов. Зачастую присутствует судорожный синдром (по типу джексоновкой эпилепсии или с генерализованными эпиприступами), характеризующийся отсутствием специфической ЭЭГ-картины.
Вариативность сочетаний различных симптомокомплексов настолько выражена, что не позволяет выделить типичный вариант течения болезни Шильдера. В ряде случаев клиника сходна с прогредиентным вариантом рассеянного склероза, в других - имеет псевдотуморозный характер, в третьих — напоминает психиатрическую патологию. В последнем случае пациенты могут проходить лечение у психиатра вплоть до развития явной неврологической симптоматики.
Диагностика лейкоэнцефалита Шильдера
Прижизненно диагностировать лейкоэнцефалит Шильдера весьма затруднительно. Эта задача требует от невролога тщательного сопоставления анамнестических, клинических и томографических данных, внимательного проведения дифдиагностики со схожими заболеваниями. С целью обследования зрительного и слухового анализаторов к консультациям могут привлекаться офтальмолог и отоларинголог.
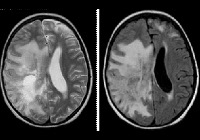
Электроэнцефалография выявляет признаки диффузного церебрального поражения: снижение альфа-активности и дезорганизацию ритма; зачастую определяется эпилептиформная активность. При исследовании цереброспинальной жидкости обнаруживается повышение уровня гамма-глобулина на фоне снижения удельного веса альбуминовой фракции. Наиболее информативным способом инструментальной диагностики выступает МРТ головного мозга. Болезнь Шильдера подтверждает наличие как минимум одного большого или пары сливных очагов демиелинизации в белом церебральном веществе.
Для установления окончательного диагноза многие неврологи руководствуются критериями C.M. Poser 1985 г.: наличие по данным МРТ 1-2 округлых зон демиелинизации величиной не менее 2х3 см; отсутствие патологии надпочечников; исключение любой иной церебральной патологии (внутримозговой опухоли, рассеянного энцефаломиелита, инсульта и пр.); соответствие норме уровня жирных кислот в сыворотке крови; выявление на аутопсии зон диффузного хронического склероза. В некоторых случаях отличить лейкоэнцефалит Шильдера от лейкодистрофии позволяют лишь гистологические исследования церебральных тканей пораженной зоны.
Лечение и прогноз лейкоэнцефалита Шильдера
Отсутствие ясных представлений об этиопатогенезе болезни Шильдера пока не позволило разработать более или менее эффективные методы ее лечения. Отмечен некоторый эффект глюкокортикостероидной терапии, в связи с чем многим пациентам назначают метилпреднизолон, вначале парентерально в ударной дозе, а затем внутрь с постепенным снижением дозы. Параллельно проводится курс нейропротекторной, антиоксидантной и сосудистой терапии, при необходимости назначаются антиконвульсантное лечение (карбамазепин, диазепам), миорелаксанты (амантадин, толперизон, амидин), противоотечные мероприятия (фуросемид, ацетазоламид, магния сульфат), психотропные фармпрепараты.
Своевременно начатое лечение способно лишь несколько задержать прогрессирование патологии. Однако, не смотря на его проведение, все пациенты погибают. Время наступления летального исхода варьирует от нескольких месяцев до 3 лет с момента дебюта лейкоэнцефалита.
Демиелинизирующие заболевания – это большая группа болезней, при которых разрушаются миелиновые оболочки структур центральной и периферической нервной системы. Они имеют мультифакториальную природу, возникают при сочетании отягощенной наследственности и внешних факторов риска. Самые распространенные нозологии: рассеянный склероз, разные клинические формы рассеянного энцефаломиелита и полинейропатий. Диагностика демиелинизирующих патологий требует проведения МРТ, нейрофизиологических и иммунологических исследований. Лечение включает гормонотерапию, иммуносупрессию, экстракорпоральную гемокоррекцию, мультидисциплинарную реабилитацию.
МКБ-10


Общие сведения
Причины
Общепринята мультифакториальная теория демиелинизирующих заболеваний, согласно которой они возникают при сочетании внешних и внутренних провоцирующих факторов. Большинство патологий связаны с наследственными причинами. Лучше всего эти закономерности изучены у больных рассеянным склерозом (РС), за развитие которого отвечают как минимум 2 гена из комплекса HLA. К экзогенным факторам риска относятся следующие:
- Инфекции. Специфический возбудитель рассеянного энцефаломиелита – вирус ОРЭМ. Зачастую демиелинизацию провоцируют вирусы кори, краснухи, инфекционного мононуклеоза. Особого внимания заслуживает ВИЧ-инфекция: энцефалопатия и деменция развиваются у 15-20% больных на стадии СПИДа.
- Метаболические нарушения. Демиелинизирующие процессы по типу миелинолиза развиваются при накоплении продуктов азотистого обмена на фоне ХПН, декомпенсированном сахарном диабете, патологии цикла обмена мочевины.
- Церебральная ишемия. Поражение миелиновых оболочек провоцируется эпизодами нарушения мозгового кровообращения. Они характерны для людей с осложненным течением артериальной гипертензии, повторными гипертензивными кризами, тяжелыми аритмиями.
- Интоксикация. В развитии демиелинизирующих заболеваний играют роль отравления химическими растворителями, лакокрасочной продукцией, угарным газом. К провоцирующим факторам относят передозировки лекарственных препаратов, которые влияют на регуляцию кардиореспираторной деятельности и вызывают гипоксию.
- Вакцинация. Описаны единичные случаи поражения ЦНС после проведения иммунизации АКДС и КПК, при факультативной вакцинации против гриппа и бешенства. Такие патологии вызваны индивидуальной реакцией организма на прививку и чаще развиваются у пациентов с отягощенной наследственностью.
Патогенез
Миелин представляет собой наружную оболочку нервных волокон и состоит из нескольких слоев плазмолеммы. Он обеспечивает электроизоляцию и питание нервов, чтобы импульсы могли быстро достигать разных структур нервной системы. Демиелинизация – патологический процесс утраты миелина при относительной сохранности аксонов. Ее следует отличать от миелинопатий – заболевания, при котором нарушаются первичные процессы образования миелиновых оболочек.
В зависимости от типа повреждения выделяют 4 вида демиелинизации: дизиммуновоспалительная, вирусная, метаболическая и гипоксически-ишемическая. Дизиммуновоспалительные формы встречаются при рассеянном склерозе и близких к нему патологиях. Она проявляется избирательной очаговой деструкцией миелина, появлением липофитов и пролиферацией микроглиоцитов. При этом большинство аксонов в ЦНС продолжают функционировать.
Вирусные демиелинизирующие заболевания развиваются при накоплении в нервной ткани патогенов, которые постепенно разрушают миелиновые оболочки. Гипоксически-ишемическое повреждение сопряжено с хроническими эпизодами гипоперфузии мозга либо с поражением ЦНС нейротоксическими агентами. Метаболическая демиелинизация может быть связана с резкими колебаниями уровня натрия. В этом случае развивается центральный понтинный миелинолиз.
Классификация
С учетом этиопатогенеза демиелинизирующие заболевания подразделяются на первичные, которые возникают без четкой причины под влиянием аутоиммунных механизмов, и вторичные – результат повреждающего действия вирусных или токсических агентов. В практической неврологии широко применяется классификация по локализации поражения и клиническому течению. Согласно ей, выделяют такие варианты демиелинизирующих болезней:
1. Поражения ЦНС. В этом случае повреждение локализовано в белом веществе головного и спинного мозга. Заболевания отличаются тяжелым течением, неуклонным прогрессированием и нарушением всех неврологических функций. По скорости развития они подразделяются на подгруппы:
- Острые. К этой категории относят первичный рассеянный энцефаломиелит и его отдельные формы: оптикомиелит, диссеминированный миелит, полиоэнцефалит. Острое течение типично для параинфекционных и вакцинальных энцефалопатий.
- Подострые. Такой тип течения характерен для рассеянного склероза, который проявляется в виде цереброспинальной, церебральной, оптической и других клинических форм.
- Хронические. В эту группу демиелинизирующих заболеваний входят энцефалиты Даусона, Петте-Деринга, диффузный лейкоэнцефалит Шильдера.
2. Поражение периферических нервов. Чаще всего диагностируется первичный полирадикулоневрит при болезни Гийена-Барре. Также к группе периферических демиелинизирующих патологий относят инфекционные и токсические нейропатии, диабетическую полинейропатию.
Симптомы демиелинизирующих заболеваний
Рассеянный склероз
Дебют происходит в молодом возрасте. Начальные симптомы представлены парестезиями в одной или нескольких конечностях, мышечной слабостью в руках и ногах, периодическими нарушениями зрения. Пациенты отмечают легкую дискоординацию движений, неустойчивость походки. Иногда в начале болезни проявляются расстройства функций тазовых органов: недержание мочи, частые позывы на мочеиспускание.
По мере прогрессирования рассеянного склероза возникают парезы или параличи конечностей, нарушаются функции черепно-мозговых нервов (ЧМН), усиливаются поражения чувствительных нервных волокон. Присоединяется спастичность мышц, которая усиливается при вертикализации пациента и во время ходьбы. Для РС типичен синдром диссоциации: несоответствие между поражениями внутренних органов и клиническими симптомами.
Острый рассеянный энцефаломиелит
Клинические проявления демиелинизирующего заболевания соответствуют энцефалопатии. Патология манифестирует нарушениями сознания разной степени тяжести – от оглушенности до комы. Пациентов беспокоят сильные головные боли, тошнота, рвота, которая не приносит облегчение. Неврологическая симптоматика достигает максимума в течение нескольких дней, из-за чего больные госпитализируются в отделение интенсивной терапии.
Очаговые проявления вариабельны и зависят от локализации поражения. Демиелинизирующая патология проявляется нарушениями координации, параличами половины дела, нарушениями зрения, речи и других функций, которые контролируются ЧМН. До 35% случаев сопровождается эпилептическими приступами, около 25% пациентов страдают от корешкового болевого синдрома, дисфункции тазовых органов.
Синдром Гийена-Барре
Главным признаком заболевания является симметричные мышечная слабость, которая начинается в ногах и постепенно распространяется на мышцы туловища, верхних конечностей, лица и шеи. Она сопровождается шаткостью походки, покалыванием в руках и ногах, болями в спине. В процесс вовлекаются мышцы, отвечающие за глотание и артикуляцию, поэтому развивается дисфагия, дизартрия. При параличе дыхательной мускулатуры возможна асфиксия.
Другие виды полинейропатий
Повреждения периферических нервов проявляются сочетанием моторных, сенсорных и вегетативных симптомов. Большинство случаев демиелинизирующего заболевания начинается с мышечной слабости в дистальных отделах конечностей, которая постепенно распространяется на вышележащие отделы тела и может достигать дыхательной мускулатуры. Характерно угнетение или полное отсутствие сухожильных рефлексов.
Осложнения
Демиелинизирующие болезни сопровождаются неврологическим дефицитом, который неумолимо прогрессирует. На поздних стадиях пациенту устанавливают группу инвалидности из-за двигательных или когнитивных нарушений. При остро протекающих заболеваниях (концентрическом склерозе, рассеянном энцефаломиелите) летальный исход возможен в первые месяцы. Диффузно-диссеминированный склероз завершается смертью через 3-7 лет от появления симптоматики.
Для демиелинизации характерно тотальное поражение мышц, поэтому как минимум у трети больных развивается дыхательная недостаточность, нарушение пережевывания и глотания пищи, расстройства речевой функции. Особенно тяжело протекает бульбарный синдром, вызванный поражением (ЧМН). Присоединяются расстройства вегетативной нервной системы, которые проявляются аритмиями, колебаниями артериального давления. нарушениями перистальтики и работы тазовых органов.
Диагностика
Для выявления демиелинизирующих заболеваний пациенту требуется полное обследование у врача-невролога. На первичной консультации большое внимание уделяется сбору анамнеза, поскольку у 80% людей выявляются факторы риска в виде перенесенных инфекций, ятрогенных вмешательств, интоксикации и прочих экзогенных вредностей. Осмотр дополняется оценкой неврологического статуса. Диагностическая программа состоит из следующих методов:
- Магнитно-резонансная томография. Нейровизуализация головного и спинного мозга является основным методом для диагностики демиелинизации в ЦНС, определения ее топографии и размеров. Внимание врачей привлекает сочетание накапливающих и не накапливающих контрастное вещество очагов в рамках одного МРТ-снимка.
- Нейрофизиологическая диагностика. При судорожном синдроме обязательно проводят классическую электроэнцефалографию и ЭЭГ с депривацией сна для выявления эпилептиформной активности. Признаки патологии периферических нервов требуется выполнение электронейромиографии, которая определяет локализацию патологии и скорость прохождения нервных импульсов.
- Иммунологические анализы. Обязательным при РС считается исследование крови и ликвора на олигоклональные антитела IgG. Чтобы определить возможные провоцирующие факторы, проводится анализ на антитела к нативной ДНК, кардиолипину, волчаночному антикоагулянту. Отличить РС от оптиконевромиелита позволяет исследование на антитела к аквапорину-4.
- Дополнительные методы. Базовая диагностическая программа включает общие анализы крови и мочи, расширенный биохимический анализ крови, определение острофазовых показателей. Для исключения хронических инфекций выполняются серологические и молекулярно-генетические реакции.
- Консультации специалистов. Зрительные нарушения требуют консультации офтальмолога, проведения офтальмоскопии, биомикроскопии глаза и визометрии. При снижении слуха пациента направляют на консультацию к отоларингологу с обязательным проведением аудиометрии, исследованием слуховых вызванных потенциалов.
Дифференциальная диагностика
Дифференциальная диагностика демиелинизирующих процессов сложна из-за разнообразия клинической картины, отсутствия четких клинико-морфологических критериев. При обследовании исключают вирусные и бактериальные энцефалиты, системные болезни соединительной ткани, паранеопластический синдром. В сложных случаях симптоматику дифференцируют с проявлениями митохондриальных заболеваний, для чего назначают биопсию мышц и ДНК-диагностику.
Лечение демиелинизирующих заболеваний
Консервативная терапия
Схема лечения подбирается индивидуально для каждого больного с учетом вида заболевания, его стадии, степени тяжести и клинических особенностей. Пациенты с умеренными проявлениями неврологического дефицита и стабильным развитием болезни проходят лечение в домашних условиях под наблюдением врача. При более тяжелых формах требуется госпитализация в неврологический стационар или реанимационное отделение. При демиелинизирующих заболеваниях показано несколько направлений консервативной терапии:
- Иммуносупрессия. Поскольку патология носит аутоиммунный характер, для купирования симптоматики назначают лечение глюкокортикостероидами. Для быстрого купирования обострения показана гормональная пульс-терапия с парентеральным введением лекарств. При их неэффективности применяются цитостатики, интерфероны, моноклональные антитела.
- Коррекция неврологических симптомов. Для ликвидации мышечной спастичности применяются миорелаксанты центрального действия, антиконвульсанты. Чтобы уменьшить координационные нарушения, используют препараты против системного головокружения. Коррекцию психоэмоционального статуса проводят антидепрессантами и анксиолитиками.
- Экстракорпоральные методы. Для удаления циркулирующих антител и иммунных комплексов проводят каскадную фильтрацию плазмы, криоаферез, лимфоцитаферез и другие методы гемоккоррекции. Терапия ускоряет наступление ремиссии и увеличивает ее продолжительность.
При вторичных формах демиелинизирующих процессов, связанных с конкретным этиологическим фактором, по возможности устраняют первопричину. Пациентам назначают противовирусную или антибактериальную терапию нейроинфекций, рациональную гипогликемическую терапию, медикаментозные и экстракорпоральные методы лечения ХПН. При токсических формах полинейропатии необходимо прекратить контакт с ядовитым веществом и ввести соответствующие антидоты.
Экспериментальное лечение
Ведутся клинические испытания блокатора ионов кальция (4-амидопирина) для купирования симптоматики демиелинизирующих процессов в ЦНС. Доказано, что препарат ускоряет проводимость по миелиновым нервным волокнам и уменьшает явления неврологического дефицита. Он действует на кальциевые каналы аксолеммы волокон, благодаря чему регулирует потенциал действия.
В 2017 году группа американских ученых представила уникальный метод генной терапии, который основан на подавлении активности иммунных клеток и ликвидации аутоиммунных повреждений миелина. Исследователи создали безопасный вирус с генетическим кодом MOG, который встраивается в ДНК печени и снижает агрессию Т-киллеров на головной мозг. Терапия находится в стадии разработки и требует длительной подготовки к клиническим исследованиям.
Реабилитация
Пациентам требуется комплексный уход и медико-социальная реабилитация. Эти меры направлены на повышение качества жизни, нормализацию физического и интеллектуального функционирования человека. Лечебная физкультура (ЛФК) улучшает силу скелетной мускулатуры, тренирует сердечно-сосудистую и дыхательную системы. Рекомендованы когнитивные тренировки, занятия с логопедом и клиническим психологом.
Прогноз и профилактика
Несмотря на усовершенствование знаний об этиопатогенезе и возможностях лечения, демиелинизирующие болезни пока представляют неразрешимую проблему для неврологии. Комплексная терапия замедляет или останавливает их прогрессирование, однако методы полного излечения не разработаны. Осторожный оптимизм внушают направления иммунотерапии и генной терапии, которые влияют на первопричину развития заболеваний.
Эффективные меры первичной профилактики отсутствуют. Чтобы снизить риск активации аутоиммунных процессов, пациентам с генетическими факторами риска рекомендуется избегать токсических воздействий, нейроинфекций, полипрагмазии лекарственных препаратов. Необходим рациональный подход к плановой вакцинации, которая предупреждает корь, краснуху и другие инфекции, выступающие триггерами демиелинизирующей болезни.
1. Неврология. Национальное руководство/ под ред. Е.И. Гусева, А.Н. Коновалова, В.И. Скворцовой. – 2018.
2. Редкие демиелинизирующие заболевания центральной нервной системы/ Т.Е. Шмидт// Неврологический журнал. – 2016. – №. 5.
3. Дифференциальная диагностика миелитов при демиелинизирующих заболеваниях/ И.С. Бакулин// Нервные болезни. – 2015. – №. 4.
4. Демиелинизирующие заболевания/ Ю.И. Стаднюк, Д.С. Лезина, О.В. Воробьева// Лечение заболеваний нервной системы. – 2012. – №2.
Лейкоэнцефалит (leukoencephalitis; греч. leukos белый + enkephalos головной мозг + -itis) — воспалительно-дистрофическое поражение белого вещества головного мозга. Лейкоэнцефалит относятся к демиелинизирующим заболеваниям (см.).
Содержание
Этиология и патогенез
Предполагается, что Лейкоэнцефалит являются заболеваниями инфекционно-аллергической природы. Дискутируется роль миксовирусов, вирусов кори, бешенства и Herpes zoster как пусковых факторов гиперергического аутоиммунного процесса.
Классификация
Патологическая анатомия

Рис. 1. Фронтальный срез головного мозга больного, умершего от геморрагического лейкоэнцефалита: белое вещество справа более отечное, усеяно мелкими петехиальными кровоизлияниями (указаны стрелками), прилежащие участки коры нечетко очерчены.

Рис. 2. Микропрепарат ткани белого вещества мозга больного, умершего от геморрагического лейкоэнцефалита: стенка кровеносного сосуда (1) инфильтрирована и окружающая нервная ткань пропитана полиморфно-ядерными лейкоцитами (2).
Макроскопическое исследование мозга при Л. выявляет расширение борозд и атрофию извилин. На срезе полушарий определяются различного размера участки деструкции и демиелинизации во всех отделах мозга, преимущественно в белом веществе, но захватывающие различные участки серого вещества коры (рис. 1). В наиболее пораженных отделах мозг имеет губчатую консистенцию, желудочки мозга умеренно расширены.
Гистол, картина характеризуется диффузной подострой воспалительной реакцией с периваскулярной инфильтрацией лимфоцитами и плазмоцитами и очаговой демиелинизацией (рис. 2). Воспалительные изменения преимущественно локализованы в белом веществе мозга, иногда в коре, подкорковых ганглиях, мозговых оболочках. Разрушается нормально сформированный миелин (миелинокластический тип поражения). Степень демиелинизации и деструкции нервной ткани варьирует в различных очагах. Отдельные мелкие очаги могут сливаться. У краев очага демиелинизации олигодендроциты увеличены, содержат амфофильные включения, в более пораженных участках они полностью исчезают. Кроме того, встречается много больших причудливой формы астроцитов с гиперхроматическими многодольчатыми или несколькими ядрами. Аксоны остаются относительно сохранными на ранних стадиях процесса, позднее в них могут быть дистрофические изменения. Нейроны коры полушарий большого мозга могут содержать включения двух типов: сферические частицы диам. 30—40 мкм и продолговатые, или тубулярные, структуры несколько меньшего диаметра. Включения чаще встречаются при небольшой длительности заболевания. Гистохимические исследования обнаруживают во включениях большое количество белка. В большинстве случаев находят пролиферативную реакцию глии. Глиоз может быть мелкоузелковый или в виде крупных очагов (псевдоопухоль). Диффузное разрастание волокнистой глии приводит иногда к уплотнению мозгового вещества, так что мозг на разрезе имеет хрящевидную консистенцию. Стенки артерий и вен утолщены, с избытком ретикулярных волокон в адвентиции.
Клиническая картина
Нервно-психические нарушения являются наиболее ранним проявлением заболевания. Вначале отмечаются жалобы на повышенную утомляемость, вялость, раздражительность, неустойчивость настроения. Постепенно круг нервно-психических расстройств расширяется. Появляется злобность, эффективность, жадность, эгоистичность, жестокость, недисциплинированность, инертность мышления. Больные часто совершают немотивированные поступки, теряют навыки опрятности.
Постоянным признаком заболевания являются судороги (см.). Они могут появляться на разных стадиях болезни. Наиболее характерны малые и абортивные судорожные припадки, реже генерализованные большие припадки. В поздней стадии заболевания развиваются трофические и вегетативные расстройства: кахексия, пролежни, нарушения терморегуляции, профузный пот и т. д. В терминальной стадии больные обездвижены, иногда наблюдается децеребрационная ригидность (см.).
Течение Лейкоэнцефалита может быть неуклонно прогрессирующим или ремиттирующим. В последнем случае клин, картина может напоминать рассеянный склероз (см.).
При электроэнцефалографии регистрируют периодическую пароксизмальную активность с интервалом 5 — 15 сек. одновременно в большинстве отведений в виде медленных (1—2 в 1 сек.) высоковольтажных волн.
В крови определяется лейкоцитоз, повышение фракции гамма-глобулина, обычно повышен титр антител к коревому вирусу или к миксовирусам (вирус jc, sv-40).
В цереброспинальной жидкости в большинстве случаев не наблюдается цитоза и увеличения содержания белка. Однако при электрофоретическом исследовании белков обнаруживают, что гамма-глобулин составляет до 40 и более процентов от общего количества белка, а фракция альбумина снижена. Коллоидные реакции дают максимальную флоккуляцию в первых пробирках (паралитический тип реакции Ланге).
Лечение
Лечение должно быть комплексным. Показана гормональная и симптоматическая терапия. Положительный эффект получают при назначении кортикостероидов. Лечение глюкокортикоидами (преднизолоном) следует начинать в ранней стадии патологического процесса с учетом ритма гормональной деятельности надпочечников. Гормональная терапия дополняется противоаллергическими (димедрол, пипольфен, супрастин, диазолин) и противосудорожными препаратами. Показаны препараты, снижающие мышечный тонус (мидокалм, амедин, мидантан, циклодол и др.), витамины группы В и другие симптоматические средства. Применение активной терапии может задерживать течение болезни и способствовать ремиссиям на несколько лет.
Прогноз
При неуклонно прогрессирующем течении больные погибают через 2—12 мес. после появления первых симптомов. При ремиттирующем течении заболевание длится до 3 лет и более, а ремиссии могут продолжаться от нескольких месяцев до нескольких лет, в течение которых симптомы заболевания почти или полностью отсутствуют.
Особенности отдельных форм лейкоэнцефалита
Подострый склерозирующий лейкоэнцефалит Ван-Богарта. При патоморфол, исследовании мозга больных с этой формой Л., как правило, обнаруживаются внутриклеточные включения. Степень поражения уменьшается в направлении от коры к филогенетически более древним образованиям, но чаще, чем при других формах, поражается ствол и спинной мозг.
Клинической особенностью этой формы Лейкоэнцефалита является раннее проявление и преобладание экстрапирамидных нарушений (Гиперкинетическая форма), к к-рым лишь на поздних этапах присоединяются пирамидные симптомы. Эпилептические припадки не характерны.
Периаксиальный диффузный лейкоэнцефалит Шильдера. Патоморфол, особенностью по сравнению с другими Л. и рассеянным склерозом является относительно ранняя дистрофия аксонов. Эта форма отличается от предыдущей преобладанием пирамидных симптомов и частыми эпилептическими припадками. Обычно наблюдаются большие припадки. Характерно развитие ретробульбарного неврита зрительных нервов или центральной формы слепоты, связанной с демиелинизацией затылочных долей (см. Шильдера болезнь).
Острый геморрагический лейкоэнцефалит. По клин, и патоморфол, признакам эта форма Л. сходна с вирусными и поствакцинальными энцефалитами. При патологоанатомическом исследовании выявляют отек мозга, на срезах в веществе мозга — большие очаги мягкой розовато-серой или желтоватой окраски с множественными точечными кровоизлияниями. Гистол. картина характеризуется фибринозным некрозом стенок мелких сосудов, в основном венул, окруженных экссудатом фибрина, воспалительными клетками и кольцевидными геморрагическими зонами. В этих же периваскулярных зонах— демиелинизация с умеренной или выраженной деструкцией аксонов. На самых ранних стадиях периваскулярные инфильтраты представлены гл. обр. нейтрофилами, однако в более старых очагах находят много лимфоцитов и плазмоцитов.
Клиника острого геморрагического Лейкоэнцефалита характеризуется чрезвычайно острым началом, молниеносным нарастанием тяжести симптомов поражения мозга. Заболевают лица обоего пола в возрасте от 20 до 40 лет. Длительность течения от 2 дней до 2 нед. Развернутой клин, картине предшествуют катаральные явления в зеве, лихорадка с лейкоцитозом в периферической крови. Через 2—4 дня появляется головная боль, ригидность мышц шеи, нарушается сознание, иногда развивается кома. Характерны фокальные или генерализованные судороги, двигательные нарушения в виде геми- или тетраплегии, псевдобульбарный паралич. На глазном дне — отек диска (соска) зрительного нерва. Редко наблюдаются подострые и хронические формы. С помощью ЭЭГ и артериографии могут быть обнаружены фокальные изменения. В цереброспинальной жидкости — выраженный плеоцитоз за счет полиморфно-ядерных лейкоцитов, встречаются также лимфоциты; содержание белка повышено до 1 г/л и более; часто выявляется ксантохромия цереброспинальной жидкости, микроскопически можно обнаружить единичные эритроциты.
Исход обычно летальный.
Библиография: Маркова Е. Д. и др. Клинико-анатомические и вирусологические данные в случае подострого склерозирующего панэнцефалита, Журн, невропат. и психиат., т. 77, № 7, с. 100 7, 1977, библиогр.; Цукер М. Б. Клиническая невропатология детского возраста, М., 1978, библиогр.; Чумаков М. Вирусологические аспекты изучения этиологии некоторых хронических заболеваний нервной системы (подострый склерозирующий панэнцефалит, вилюйский энцефаломиелит, множественный склероз), в кн.: Демиелинизирующие заболевания нервн. системы в Эксперим, и клин., под ред. А. И. Булыгина, с. 79, Минск, 1975; Balakоva H., Kvicalа V. Radiologicke a scintigraficke nalezy u akutnich zanetlivech procesii CNS, Cs. Neurol. Neurochir., sv. 40, s. 350, 1977; Gilroy J. a. Meyer J. S. Medical neurology, N. Y., 1975; Lhermitte F. Les leucoencephalites, P., 1950, bibliogr.; Pette H. u. Doring G. Uber einheimische Panencephalomyelitis vom charakter der Encephalitis japonica, Dtsch. Z. Nervenheilk., Bd 149, S. 7, 1939.
Facebook Если у вас не работает этот способ авторизации, сконвертируйте свой аккаунт по ссылке ВКонтакте Google RAMBLER&Co ID
Авторизуясь в LiveJournal с помощью стороннего сервиса вы принимаете условия Пользовательского соглашения LiveJournal
Болезнь Шильдера

Болезнь Шильдера, или диффузный периаксиальный энцефалит Шильдера (ДПЭ) [син.: подострый склерозирующий панэнцефалит, периаксиальный диффузный лейкоэнцефалит, симметричный интерглобулярный склероз] - это редкое, неуклонно прогрессирующее заболевание нервной системы, характеризующееся формированием патологических обширных очагов демиелинизации в белом веществе обоих полушарий головного мозга, часто асимметричных, с четко очерченными и заостренными краями. Также участки демиелинизации могут первично встречаться в мозжечке и стволе мозга.
Некоторые авторы описывают случаи, когда наряду с основными, большими очагами, также встречаются участки несколько меньшего размера, округлой формы, напоминающие бляшковидные участки демиелинизации при рассеянном склерозе. Их появление более характерно для заболевания, начавшегося в подростковом и зрелом возрасте. При патогистологическом исследовании выявляются участки фибриллярного глиоза с гигантскими многоядерными астроцитами и периваскулярная инфильтрация плазматическими клетками.
Этиология ДПЭ остается неизвестной. Вопрос возрастного дебюта заболевания остается спорным и имеет неоднозначные данные. Зарубежные авторы в своих наблюдениях подчеркивают, что для ДПЭ характерно начало в детском возрасте (7 - 12 лет). Однако ряд отечественных ученых настаивают, что частота заболеваемости не зависит от возрастной категории и отмечаются одинаково часто как у детей, так и у взрослых.
Клиническая картина болезни ДПЭ полиморфна и неспецифична. Выделяют следующие основные группы симптомов: [ 1 ] психические расстройства, сопровождающиеся нарушением поведения по апато-абулическому типу, а также когнитивным снижением, вплоть до тотальной деменции; [ 2 ] поражение черепно-мозговых нервов (глухота, офтальмоплегия, парез лицевого нерва, бульбарный синдром, оптический неврит; [ 3 ] поражение мозжечка (нистагм, интенционный тремор, скандированная речь, атаксия); [ 4 ] поражение зрительной коры (корковая слепота, гемианопсия); [ 5 ] судорожный синдром (чаще всего не сопровождающийся специфическими изменениями на ЭЭГ); [ 6 ] экстрапирамидные нарушения; [ 7 ] общемозговые симптомы.
Прижизненная диагностика ДПЭ остается затруднительной и требует тщательного анализа данных и дифференцирования от ряда других клинически схожих заболеваний. Основным методом диагностики является МРТ-исследование головного мозга, которое должно показать наличие одного большого или двух сливающихся между собой участков демиелинизации в белом веществе головного мозга, чаще всего расположенных перивентрикулярно, била- терально или монолатерально. ЭЭГ-данные при ДПЭ неспецифичны и проявляются в виде дезорганизации волн и снижении a-активности, что свидетельствует о диффузном поражении головного мозга. Возможное наличие латерализованных эпилептиформных разрядов (PLEDS) указывает на развитие подострого склерозирующего энцефалита, особенно если дебют заболевания приходится на детский возраст.
1 - один или два очага округлой формы, расположенные симметрично в каждом полушарии, преимущес твенно в семиовальном центре; размер очагов составляет не менее 2 × 3 см;
2 - отсутствие клинических или лабораторных данных относительно патологии надпочечников;
3 - сывороточная концентрация жирных кислот с длинной цепью в пределах физиологической нормы;
4 - отсутствие любых других поражений головного мозга, определяющихся клинически, лабораторно или инструментально;
5 - отсутствие патологии со стороны периферической нервной системы;
6 -наличие очагов диффузного хронического склероза на аутопсии.
Читайте также: