Лечение красного волосяного лишая девержи
Обновлено: 15.04.2024
ГКБ №14 им. В.Г. Короленко Департамента здравоохранения Москвы
Городская клиническая больница №14 им. В.Г. Короленко Департамента здравоохранения Москвы
ГУ МОНИКИ им. М.Ф. Владимирского
Ограниченный ювенильный тип болезни Девержи
ГКБ №14 им. В.Г. Короленко Департамента здравоохранения Москвы
ГКБ №14 им. В.Г. Короленко Департамента здравоохранения Москвы
Городская клиническая больница №14 им. В.Г. Короленко Департамента здравоохранения Москвы
ГУ МОНИКИ им. М.Ф. Владимирского
Заболевание красный волосяной лишай (КВЛ, Pityriasis rubra pilaris) известно около 200 лет. Впервые его упомянул C. Tarral (1829 г.), затем подробное описание представил M. Devergie в 1857 г., а сам термин предложил E. Besnier в 1889 г. [12, 14, 25].
Болезнь Девержи (БД) (син.: красный волосяной отрубевидный лишай, pityriasis rubra pilaris) — редкий идиопатический гиперпролиферативный дерматоз, характеризующийся фолликулярными роговыми папулами, кератодермией, различной степенью выраженности эритемой и шелушением [4, 6, 7]. Патологическая сущность заболевания связана с нарушением ороговения на фоне гиперактивности кератиноцитов, а также с дисметаболизмом витамина, А и ослаблением белковосвязывающей функции печени [3, 4, 8].
Этиология БД окончательно не установлена. На развитие болезни влияют вегетативные и гормональные дисфункции, гепатозы, инфекции, инсоляция, вакцинации и др. [1, 4, 5].
Встречается заболевание, по различным источникам, с частотой 0,03—1,3% всех болезней кожи [5, 8, 28]. Его дебют приходится на первое или пятое десятилетие жизни [4, 14, 21]. Врожденная форма болезни имеет аутосомно-доминантный тип наследования, а приобретенная (спорадическая) генетически не детерминирована [3—5].
Заболевание начинается с поражения фолликулярного аппарата. Вначале на фоне эритемы на различных участках кожного покрова появляются изолированные милиарные фолликулярно-гиперкератотические и конусовидные папулы. Скученные элементы сыпи, увеличиваясь в числе и сливаясь, образуют бляшки различных размеров [9, 20, 26]. Первичные морфологические элементы болезни — остроконечные фолликулярные папулы желтовато-оранжевого, розовато-бурого цвета, покрытые плотно прикрепленными беловатыми отрубевидными или пластинчатыми чешуйками с пушковыми волосами в центре [7, 14, 24]. Вокруг основных бляшечных высыпаний видны единичные милиарные узелки [13, 18]. Под влиянием провоцирующих факторов заболевание может обостряться, а кожный процесс принимает распространенный характер.
В 1980 г. W. Griffiths предложил классификацию КВЛ, основанную на клинической характеристике и течении заболевания [1, 3, 4, 11]. Были выделены пять клинических типов (форм):
I — классический у взрослых;
II — атипичный у взрослых;
III — классический юношеский;
IV — ограниченный ювенильный;
V — атипичный или невоидный юношеский.
БД отличается выраженным клиническим полиморфизмом.
Атипичный взрослый тип II отличается хроническим течением, продолжительными обострениями и кратковременными ремиссиями. Встречается этот тип у 5% больных Б.Д. Может начинаться с эритематозно-сквамозных и гиперкератотических очагов поражения на разгибательных поверхностях лучезапястных и локтевых суставов. В некоторых случаях первично изменяются ногтевые пластинки: они становятся тусклыми, исчерченными, с онихогрифозом. Иногда наблюдаются экзематозные поражения кожи, поредение волос на голове. Чешуйки при этой форме более крупные, ихтиозиформные. Нередко образуются различного размера сквамозные бляшки из перифолликулярных папул. Бывают также ограниченные очаги фолликулярного кератоза. Эта форма особенно резистентна к терапии.
Классический ювенильный тип III возникает на 1-м или 2-м году жизни. Клинически проявляется одинаково с классическим взрослым I типом, различается только возраст пациентов [1, 29]. От взрослого типа он отличается более частым (в 2 раза) началом на нижней половине туловища и медленным распространением. На фалангах пальцев также может возникать фолликулярный гипекератоз. По сравнению с классическим взрослым типом, клиническая картина менее выражена.
Ограниченный ювенильный тип IV, возникая в первые годы жизни, наблюдается в 25% случаев заболевания КВЛ, характеризуется приобретенными резко ограниченными очагами фолликулярного гиперкератоза и инфильтрации на коже коленных и/или локтевых суставов, реже на других участках кожного покрова. На ладонях и подошвах отмечается утолщение рогового слоя, иногда трещины. В части случаев имеет место ониходистрофия [10, 30].
Абортивный (ослабленный) V тип болезни Девержи проявляется многообразно, в частности ладонно-подошвенными гиперкератотическими бляшками, редко встречающимися фолликулярными папулами на лице, чаще — сквамозной эритемой [13, 21, 26]. Поражения слизистых редки, напоминают таковые при красном плоском лишае [13, 15]. Кожа головы, за редким исключением, не вовлекается в патологический процесс, при этом высыпания проявляются в виде беловатых чешуек, а также дистрофическим изменением волос. Иногда клиническую картину дополняет ладонно-подошвенная кератодермия [2, 5, 10]. Оттенки цвета пораженной кожи — от оранжевого до розово-красно-коричневого [3, 5, 17, 19].
Поражения кожи в детском возрасте могут существовать годами без тенденции к распространению. Сезонность в их хроническом течении отсутствует [12, 13]. Отмечаются спонтанные ремиссии и экзацербации [3]. Общее состояние не страдает. Субъективные ощущения — зуд, жжение, стягивание кожи различной интенсивности [14]. Прогноз неопределенный. Иногда наступает спонтанное разрешение в позднем подростковом возрасте. Гистологически настоящий тип имеет сходство с псориазом, но без микроабсцессов Мунро.
Вариабельность БД проявляется типичной (классической), атипичной (ограниченной) и эритродермической формами [4]. Клинически дерматоз характеризуется фолликулярным гиперкератозом, перифолликулярной эритемой, склонной переходить в эритродермию с островками непораженной кожи на ее фоне, отрубевидным шелушением, ладонно-подошвенным гиперкератозом [10, 12, 24].
Дифференциальная диагностика ограниченной формы БД проводится с себорейным дерматитом, псориазом, атопическим дерматитом, фолликулярным ихтиозом, красным плоским лишаем, вариабельной эритрокератодермией, лихеноидным парапсориазом, Т-клеточной лимфомой, ВИЧ-инфекцией, врожденной ихтиозиформной небуллезной эритродермией [1, 2, 4, 27].
Хотя патогномоничных гистологических изменений при БД нет, патоморфологическая картина имеет определенные особенности. В эпидермисе обнаруживают гранулез, фокальный паракератоз и акантоз. Может также быть вакуольная дистрофия клеток базального слоя, роговые пробки в устьях волосяных фолликулов; в дерме — умеренная инфильтрация из меланоцитов и гистоцитов, располагающаяся вокруг сосудов волосяных фолликулов [2, 4—6]. Некоторые авторы отмечают отдельные расхождения клинической и гистологической картины.
Лечение БД — непростая задача. Целесообразна комплексная терапия, включающая широкий круг лекарственных средств. Препаратами первого выбора являются системные ретиноиды и метотрексат. Применяют витамины А, Е, группы В, глюкокортикостероидные препараты (локально и системно), кератомикотики, эмоленты, 2% серно-дегтярные и салициловые мази, мази с мочевиной; гелио- и талассотерапию [2, 13, 23]. Иногда возможно спонтанное разрешение заболевания [1, 3, 5]. Примерно в 1/3 случаев дерматоз разрешается в сроки от нескольких месяцев до 5 лет [10, 13, 21]. Полезны теплые общие ванны с морской солью, крахмальные с последующим применением кератолитических мазей, фонофореза гидрокортизонового крема с добавлением аевита. Прогноз для жизни благоприятный, в отношении излечения — неопределенный [5, 22, 25].
Клинический случай
Status localis: патологический процесс имеет хронический подостро воспалительный характер, локализован на коже разгибательной поверхности локтевых и коленных суставов, ягодиц (рис. 1−3).
Рис. 1. Очаги поражения кожи коленных суставов. Представлен фолликулярными папулами, местами сливающимися в бляшки с чешуйчато-корковыми наслоениями, размером от 3,5 до 5,5 см, округло-продолговатой формы, буровато-коричневого цвета со слабым желтоватым оттенком, при пальпации плотноватой консистенции. Дермографизм красный. Рис. 2. Патологический процесс на коже локтевых суставов. Рис. 3. Редко наблюдающиеся поражения кожи ягодиц.
Проведено лечение. Наружно назначена 2% салициловая мазь, топикрем, пимафукорт в течении 1-й недели. Отмечена положительная динамика в очагах поражения. В дальнейшем на фоне терапии, включающей топические кортикостероиды, эмоленты, кремы, содержащие салициловую кислоту, мочевину, патологический кожный процесс разрешился до состояния клинической ремиссии.
Рис. 4. Участок с агранулезом, паракератозом и нейтрофилами в роговом слое по краю биоптата (окраска гематоксилином и эозином, ув. ×400). Изменения характерны для псориаза, но также нельзя исключить вторичный характер изменений (травма, присоединение вторичной инфекции). Рекомендована повторная биопсия для исключения/подтверждения псориаза. Рис. 5. Характерный для болезни Девержи морфологический признак — чередование участков орто- и парагиперкератоза в вертикальном и горизонтальном направлениях (окраска гематоксилином и эозином, ×400). В верхних отделах дермы — периваскулярые лимфогистоцитарные инфильтраты, полнокровие сосудов. Заключение: гистологически больше данных за псориаз; нельзя также исключить болезнь Девержи.
Представленное наблюдение представляет несомненный клинический интерес в связи с редкостью и многообразием клинических проявлений наблюдаемого дерматоза.
Сведения об авторах
КАК ЦИТИРОВАТЬ:
Приведены критерии диагностики красного волосяного лишая Девержи и подходы к лечению больных. Применение синтетических ретиноидов дает возможность повысить эффективность терапии таких пациентов. Однако лечение БД является длительным и всегда сопровождаетс
Красный волосяной лишай Девержи (син.: болезнь Девержи; pityriasis rubra pilaris; lichen ruber acuminatus) — гетерогенное хроническое воспалительное заболевание кожи, которое подразделяется как на наследственные формы, передающиеся аутосомно-доминантно, так и на спорадические, приобретенные. Хотя принято считать, что название и описание принадлежит Альфонсу Девержи, о первом случае сообщил в 1828 г. Клаудиус Таррал. Он отмечал изолированные чешуйчатые высыпания, пронизанные в центре волосом, при пальпации которых на поверхности кожи прощупывается очень плотная шероховатость [1–6].
Болезнь Девержи (БД) — редкое заболевание, составляющее 0,03% всех болезней кожи. Этиология и патогенез дерматоза до сих пор до конца не изучены, мнения современных авторов расходятся. Теория наследственной предрасположенности занимает главенствующую роль, хотя существуют и другие теории [3, 5]. Иммуноморфологическое исследование клеток воспалительного инфильтрата у больных БД характеризовалось повышенным количеством клеток с экспрессией маркеров, принимающих участие в развитии воспалительной иммунной реакции по механизму гиперчувствительности замедленного типа. Это позволяет отнести заболевание к хроническим дерматозам, характеризующимся гиперпролиферацией кератиноцитов, опосредованной Т-клетками [7]. По данным К. Н. Суворовой и соавт. (1996), начало заболевания может отмечаться во всех возрастных периодах от 5 до 72 лет. Течение заболевания хроническое, иногда десятилетиями, сезонности не отмечается [8].
В 1980 г. Гриффитс описал 5 клинических форм заболевания, которыми достаточно долго пользовались врачи:
- Классический взрослый тип заболевания.
- Атипичный взрослый тип.
- Классический ювенильный тип.
- Ограниченный ювенильный тип.
- Атипичный ювенильный тип.
Однако в современной дерматологии принято считать, что классический взрослый тип и классический ювенильный типы заболевания имеют идентичные клинические проявления и различаются только возрастом пациентов. По этой причине в настоящее время классификация БД включает три клинические формы:
- Классический тип.
- Ограниченный ювенильный тип.
- Тип, связанный с ВИЧ-инфекцией [6].
Клиническая диагностика заболевания основывается на характерных признаках:
Однако довольно часто высыпания при БД напоминают псориазиформные элементы, а наличие диффузной эритродермии придает схожесть дерматоза с псориазом и токсикодермией. Псориаз и БД объединяют не только сходство клинических проявлений, но и механизмы развития воспалительного процесса. Результаты гистологического и иммуноморфологического исследования, проведенного О. Р. Катуниной (2005), свидетельствуют об общем патогенетическом пути развития воспалительного процесса в коже при псориазе и БД. В обоих случаях происходит активация антигенпредставляющих клеток, гиперпролиферация кератиноцитов, которые, приобретая черты иммуноцитов, способствуют поддержанию патологического процесса [7].
Диагностика
Следующим очень важным симптомом БД является характерная окраска кожи — кирпично-красная или морковная, описанная многочисленными авторами [3–10]. Нами отмечено, что морковный оттенок кожи имели 7 пациентов (30,4%), а у остальных наблюдалась кирпично-красная окраска (n = 16; 69,6%). Однако ладонно-подошвенный гиперкератоз, развившийся у 19 (82,6%) из наблюдаемых нами больных, во всех случаях давал желтовато-морковную окраску кожи.
Хотя поражение ногтевых пластинок среди обследованных больных диагностировалось только у 18 (78,2%) пациентов, хочется отметить, что выраженность данного признака, скорее всего, зависит от давности заболевания, поскольку скорость роста ногтевой пластинки значительно медленнее, чем развитие патологического процесса. Это предположение подтвердили наши наблюдения: у больных, имеющих поражение ногтевой пластинки, длительность заболевания составила от 4 до 10 месяцев. Изменение ногтевой пластинки характеризовалось подногтевым гиперкератозом, изменением цвета, продольной и поперечной исчерченностью. Однако онихолизис, характерный для псориаза, не отмечен ни у одного пациента.
Как уже упоминалось выше, у большинства наблюдаемых нами пациентов с БД (n = 21; 91,2%) первоначально правильный диагноз не был установлен. Пациенты получали гипосенсибилизирующие и антигистаминные препараты, а более половины из них (n = 15; 65,2%) — пролонгированные стероиды, но, несмотря на интенсивную терапию, положительной динамики в лечении не отмечалось. Данный факт еще раз подтверждает, что БД резистентна к обычной терапии. Резюмируя вышесказанное, можно выделить основные дифференциально-диагностические признаки, позволяющие отличить красный волосяной лишай от псориаза:
- хорошее общее состояние больных БД сохраняется даже при диффузном поражении кожного покрова, в отличие от псориаза;
- характерным признаком является наличие островков здоровой кожи;
- первичным элементом является остиофолликулярная папула;
- кирпично-красная или морковная окраска кожи;
- отсутствие инфильтрации, лихенификации и обильного крупно-пластинчатого шелушения, свойственных псориазу;
- онихолизис, характерный для псориаза, наблюдается очень редко;
- ладонно-подошвенный гиперкератоз без инфильтрации, дающий желтовато-морковный оттенок кожи;
- торпидность к гипосенсебилизирующей, антигистаминной и гормональной терапии.
Лечение
В настоящее время препаратами, воздействующими на процессы кератинизации, являются ретиноиды. Впервые в 1930 г. Moore синтезировал ретинол из каротиноидов и начал изучать его действие на организм. Сегодня доказано, что витамин А участвует в регуляции и пролиферации многих типов клеток с момента эмбриональной закладки и в течение всей жизни. Наиболее эффективным среди всех синтетических ретиноидов в лечении БД является Неотигазон в дозе 0,5–0,7 мг/кг/сутки [4–6, 10].
Неотигазон (ацитретин 10 мг, 25 мг) обладает выраженной липофильностью и легко проникает в ткани. Нужно учитывать, что из-за индивидуальных различий всасывания и скорости метаболизма ацитретина дозу нужно подбирать индивидуально. Начальная суточная доза составляет 25 или 30 мг в сутки в течение 2–4 недель. Капсулы лучше принимать один раз в сутки вечером во время еды или с молоком. Как правило, терапевтический эффект достигается при суточной дозе 30 мг. В некоторых случаях бывает необходимо увеличить дозу до максимальной, равной 75 мг/сутки. Однако не следует рассчитывать на быстрый клинический эффект от проводимой терапии при лечении БД, особенно у взрослых. Наше мнение совпадает с мнением зарубежных авторов, которые считают, что симптоматическое улучшение при БД наступает в течение 1 месяца, но значительное улучшение и, может быть, разрешение процесса возможно в течение 4–6 месяцев, а по некоторым данным средняя продолжительность терапии составляет около 4 лет [6]. Наши наблюдения за пациентами с БД установили средний срок терапии 9 месяцев (рис. 4).
.jpg)
Учитывая возможность развития тяжелых побочных явлений, при длительном лечении следует тщательно сопоставить возможный риск с ожидаемым терапевтическим эффектом. Противопоказаниями к применению Неотигазона являются гиперчувствительность к препарату (ацитретину или наполнителям) или к другим ретиноидам; тяжелая печеночная и почечная недостаточность; выраженная хроническая гиперлипидемия, хронический алкоголизм; желчнокаменная болезнь, хронический панкреатит; заболевания центральной нервной системы, сопровождающиеся повышением внутричерепного давления. А. А. Кубанова и соавт. (2005) указывают на противопоказания к назначению ретиноидов при наличии в анамнезе эпителиомы (в том числе семейной), чешуйчато-клеточной или базально-клеточной карциномы [10]. Неотигазон обладает сильным тератогенным действием. Риск рождения ребенка с пороками развития особенно высок, если Неотигазон принимают до или во время беременности, вне зависимости от дозы и продолжительности терапии. В литературе указываются различные временные промежутки между окончанием приема ретиноидов и беременностью, необходимые для исключения риска возникновения тератогенного эффекта. По данным различных авторов этот период составляет от 6 месяцев до 2 лет [10].
У пациентов с сахарным диабетом ретиноиды могут улучшить или ухудшить толерантность к глюкозе, поэтому на ранних этапах лечения концентрацию глюкозы в крови следует проверять чаще обычного.
Из-за риска развития гипервитаминоза А следует избегать одновременного применения витамина А и других ретиноидов. Поскольку как Неотигазон, так и тетрациклины могут вызывать повышение внутричерепного давления, их одновременное применение противопоказано. При комбинированном применении метотрексата и Неотигазона возникает риск развития гепатита, поэтому назначение одновременно этих двух препаратов также противопоказано.
Для уменьшения токсического влияния Неотигазона на организм назначают адъювантную терапию: гепатопротекторы, липотропные средства и желудочные ферменты, витамины группы В, никотиновую и пантотеновую кислоты. Для снижения риска возникновения гиперлипидемии, во время лечения следует избегать пищи, богатой жирами.
Согласно мнению различных авторов, альтернативными методами лечения могут быть Преднизолон (15–20 мг/сут), Дипроспан (2,0 в/м один раз в 10 дней № 3) и цитостатики: Проспидин (по 50–100 мг в/м ежедневно на курс 2,0–3,0 г) или Метотрексат (15 мг в/м один раз в 7 дней) [6, 11].
Наружное лечение во время приема ретиноидов принципиального значения не имеет, однако существенно улучшает общее состояние больного и повышает качество его жизни. Используются как классические мази и кремы, так и современные средства по уходу за сухой кожей различных фирм производителей. Для ликвидации массивных роговых наслоений назначаются мази с 2–5% салициловой кислотой, 10% мочевиной, 1–20% яблочной кислотой.
Кроме того, для лечения пациентов с БД применяется физиотерапия. В основном используются различные ванны: сульфидные продолжительностью 6–10 мин через день, на курс 12–15 ванн; радоновые ванны положительностью 10–20 мин через день; в домашних условиях возможно применять крахмальные ванны: 300–500 г картофельного крахмала разводят в 2–5 л холодной воды и затем вливают в ванну температурой 36–37 °С, длительность процедуры 15–20 мин через день, курс составляет 15–20 процедур. У детей продолжительность процедуры не должна превышать 15 минут, курс составляет 8–12 ванн.
До сих пор не решен вопрос о целесообразности применения ультрафиолетового облучения (УФО) у таких пациентов, мнения современных авторов противоречивы и требуют дальнейшего изучения [6, 12]. Согласно нашим наблюдениям у всех обследованных пациентов УФО или инсоляция провоцировали обострение процесса.
Благодаря достижениям отечественной и зарубежной науки в последние годы произошли существенные сдвиги в понимании клинико-генетического полиморфизма наследственной кожной патологии, значительно продвинулись познания в области патоморфологии и иммуноморфологии, что расширило возможности точной диагностики БД. Применение синтетических ретиноидов дало возможность повысить эффективность терапии этой категории больных. Однако лечение БД является длительным и всегда сопровождается развитием ряда побочных эффектов, что приводит к ограничению возможностей назначения препарата пожилым людям и пациентам с отягощенным анамнезом.
Литература
А. А. Кубанов*, доктор медицинских наук, профессор
Ю. А. Галлямова **, 1 , доктор медицинских наук, профессор
* ФГБУ ГНЦДК МЗ РФ, Москва
** ГБОУ ДПО РМАПО МЗ РФ, Москва
Abstract. Diagnostics of Devergie pityriasis rubra pilaris criteria and approaches to treatment are presented in the article. Application of synthetical retinoids allows increasing effectiveness of therapy of such patients. But treatment of Devergie disease require long time and is followed by development of several side effects that cause limitations for prescription of medicine to elderly people and to patients with compromised history.
Красный отрубевидный волосяной лишай Девержи (син.: болезнь Девержи, лишай красный остроконечный Гебры—Капоши) является редким хроническим дерматозом и составляет 0,03% от всех болезней кожи 1, 2. Встречается в любом возрасте во всех широтах и у лиц всех рас, одинаково часто у мужчин и женщин 3. По данным анализа родословных, болезнь Девержи (БД) не следует относить к моногенным дерматозам 4. Выделяют наследственную (с аутосомно-доминантным типом наследования) и приобретенную формы 1.
Первичным элементом БД является фолликулярный гиперкератоз. Эритема и шелушение могут образовывать обширные хорошо ограниченные очаги поражения. В части случаев развиваются эритродермия, ладонно-подошвенный гиперкератоз, изменение ногтей, поражение слизистой оболочки рта, глаз, лимфаденопатия. Выпадение волос наблюдается редко, хотя поражение волосистой части головы типично для этого дерматоза. Начало БД может быть постепенным или с самого начала заболевание характеризуется быстрым прогрессирующим течением и за несколько дней развивается эритродермия. При длительном поражении лица может возникнуть эктропион. Поражаются фолликулы кожи, чаще в области колен и локтей, задней половины шеи, тыльной поверхности проксимальных фаланг пальцев кистей. В период прогрессирования заболевания может наблюдаться зуд различной интенсивности, возможен феномен Кебнера. Различные факторы — ожоги (солнечные, электрические, химические), травмы головы, прививки, коклюш, скарлатина, ветряная оспа, фурункулез, ангина и др. могут ускорить течение заболевания. БД длится от нескольких месяцев и в течение всей жизни. Полные ремиссии, как и рецидивы, встречаются часто 3.
W.A. Griffiths различает 6 клинических вариантов БД 5. Первый, классический, характеризуется острым началом в виде эритематозных пятен или эритродермии с сохранением небольших островков здоровой кожи. Второй, атипический, у взрослых отличается хорошо выраженным ладонно-подошвенным гиперкератозом, поредением волос на голове и длительным течением. Третий, четвертый и пятый, юношеские варианты БД, начинаются с раннего детства или в пубертатном возрасте ограниченными очагами с фолликулярным гиперкератозом и эритемой, наблюдаются на протяжении нескольких лет и принимают хроническое течение. Шестой тип заболевания, ассоциированный с ВИЧ-инфекцией, может проявляться узловато-кистозными и пустулезно-акнеиформными элементами, трудно поддающимися стандартному лечению.
Рисунок 1. Классическая картина эритродермической формы болезни Девержи у женщины 45 лет, длительно страдающей настоящим заболеванием: гиперемия кожных покровов с оранжевым оттенком, островки непораженной кожи.
Рисунок 2. Та же больная, островки непораженной кожи при эритродермической форме болезни Девержи.
Дифференциальную диагностику эритродермической формы БД следует проводить с эритродермической формой грибовидного микоза, который развивается преимущественно у людей старше 40 лет. При этом изменения кожи стойкие, пораженная кожа красно-фиолетового оттенка, отечная и инфильтрированная, выражена генерализованная лимфаденопатия, интенсивен постоянный зуд, шелушение чаще мелкопластинчатое, на лице и волосистой части головы отрубевидное 6. Эритродермию при БД необходимо также отличать от псориатической эритродермии. С диагностической точки зрения при последнем заболевании имеют значение сохраняющиеся отдельные псориатические элементы, яркий тон пораженной кожи и обильное шелушение 6.
В лечении БД эффективны повторные курсы витамина А из расчета 150 000-200 000 ЕД?сут в течение 3—6 нед 7. Van Voorst Vader при эритродермической форме применял витамин А в суточной дозе 1 млн ЕД в течение 18 дней 8. Показаны также витамины группы B, Е, аевит. При остром течении эритродермии используют ретиноиды (изотретиноин, тигазон, этретинат — от 0,75 до 1,03—1,48 мг?кг?сут) 7, 9; кортикостероидные препараты (преднизолон по 20—40 мг?сут); при слабой эффективности ретиноидов и кортикостероидов — цитостатические средства (циклоспорин, метотрексат, азатиоприн). В ряде случаев с успехом используется ПУВА-терапия в комбинации с ретиноидами, аевитом, а также УФ-облучение в комбинации с отваром полевого хвоща внутрь 7.
Под нашим наблюдением находился пациент Х., 49 лет, с первичной эритродермической формой БД, имеющей своеобразную клиническую картину. На фоне нормальной кожи за 10 дней развилась эритродермия с обильным шелушением, зудом, гиперкератозом ладоней и подошв, полиаденией, снижением массы тела (8 кг). До этого по поводу ОРЗ, осложненного бронхитом, больной принимал бисептол, тетрациклин и гентамицин. Заболевание проявилось эритематозными пятнами на лице, туловище, а затем на всем кожном покрове. С предварительным диагнозом токсикодермии 11.10.02 больной госпитализирован в РКВД МЗ РД. При поступлении общее состояние удовлетворительное, питание пониженное. Весь кожный покров гиперемированный, отечный, с обильным шелушением (рис. 3, 4); на ладонях и подошвах кожа характерного морковного цвета с шелушением и трещинами, выраженный гиперкератоз (рис. 5, 6). Суставы ногтевых фаланг обоих указательных пальцев деформированы, свободный край ногтей утолщен (рис. 7). Волосы не выпадают. Подмышечные и паховые лимфатические узлы увеличены до 2—3 см в диаметре, в легких везикулярное дыхание. Тоны сердца приглушены, ритмичны. Живот мягкий, безболезненный. Печень не увеличена, селезенка не пальпируется. Стул и мочеиспускание в норме.
Рисунок 3. Больной Х., 49 лет. Первичная эритродермическая форма болезни Девержи. Тотальная эритродермия, лимфаденопатия паховых лимфатических узлов.
Рисунок 4. Тот же больной. Гиперкератоз, скопление корок, шелушение и деформация ногтей пальцев стопы.
Рисунок 5. Лихенификация, инфильтрация, образование трещин на коже тыльной поверхности кистей, гиперкератоз ладоней.
Рисунок 6. Гиперкератоз на подошвах.
Рисунок 7. Деформация концевых фаланг указательных пальцев, поражение ногтевых пластинок.
При обследовании в общем анализе крови от 12.10.02 гемоглобин 133 г?л, эритроциты 3,65?1012?л, лейкоциты 7,0?109?л, э. 3, с. 68, п. 2, л. 27, СОЭ 6 мм?ч; глюкоза крови 3,6 ммоль?л, билирубин общий 4,27 мкмоль?л, тимоловая проба 2 ЕД. Серологические реакции крови отрицательные. Общий анализ мочи без патологии; в кале яйца глистов не обнаружены.
С диагнозом эритродермия неясного генеза больной был направлен и госпитализирован в ГУ ЦНИКВИ МЗ и СР РФ. Приводим данные, представленные в выписке из истории болезни отделения дерматологии ЦНИКВИ.
При микроскопическом исследовании двух биоптатов кожи, проведенном в ГУ ЦНИКВИ МЗ и СР РФ с интервалом один год, выявлена идентичная картина: акантоз, гиперкератоз с шелушением, с чередующимися участками паракератоза. Роговые скопления с паракератозом в устьях волосяных промежутков в шиповатом слое с одиночными очагами акантолиза. Признаков экзоцитоза и гидропической дистрофии базальных клеток нет. В дерме — отек и довольно скудные лимфогистиоцитарные инфильтраты вокруг сосудов, стенки которых утолщены. При иммунофенотипировании обнаружено явное преобладание лимфоцитов СD45+ и СD4+ по сравнению с клетками СD8+. Заключение: убедительных данных в пользу лимфопролиферативного заболевания нет. Гистологические изменения характерны для красного волосяного лишая Девержи.
Биохимический анализ крови: протромбиновый индекс 91 (норма 90—105), АлАТ 12,1 МЕ?л (норма 10—37 МЕ?л), АсАТ 18,5 МЕ?л (норма 10—37 МЕ?л), мочевина 2,8 ммоль?л (норма 1,7—6,3 ммоль?л), холестерин 4,06 ммоль?л (норма 3,1—5,7 ммоль?л), общий белок 68,6 г?л (норма 62,0—85,0 г?л), триглицериды 0,92 ммоль?л (норма 0,6—2,3 ммоль?л), общий билирубин 10 мкмоль?л (норма 8,5—20,5 мкмоль?л), глюкоза 4,98 ммоль?л (норма 4,2—6,1 ммоль?л), креатинин 101,3 ммоль?л (норма 44—97 ммоль?л). Антитела к ВИЧ и маркеры гепатита не обнаружены.
УЗИ органов брюшной полости, а также рентгенограмма органов грудной клетки: без выраженных патологических изменений. Забрюшинные и мезентериальные лимфатические узлы не выявлены. При анализе пунктата костного мозга изменения, свидетельствующие о лимфопролиферативном заболевании, не выявлены.
Общий анализ мочи и крови в пределах нормы. Копрограмма: недостаточность желудочного пищеварения; бродильный дисбиоз (толстой кишки). Исследование кала на дисбактериоз: снижение количества лактобактерий.
Клинический диагноз: эритродермическая форма болезни Девержи.
В течение 2 мес было проведено следующее лечение:супрастин 2,0 внутримышечно (всего 10 инъекций); гемодез 400,0 внутривенно (9 вливаний); глюконат кальция 10% 10,0 внутримышечно (9 инъекций); тавегил 2,0 внутримышечно (7 инъекций); дипроспан 1,0 внутримышечно однократно; мезим форте 1 таблетка 3 раза в сутки (26 дней); диазолин 0,1 внутрь 3 раза в сутки (8 дней); альмагель 1 столовая ложка 1 раз в день (7 дней); преднизолон внутрь 20 мг?сут с постепенным снижением дозы; феназепам 0,01 внутрь на ночь (31 день); кларитин 0,1 внутрь
1 раз в день (9 дней); неотигазон внутрь 0,01 1 раз в день
(2 дня) и 0,02 1 раз в день (25 дней); лактобактерин внутрь 2 капли 3 раза в день (14 дней); кестин 0,01 внутрь на ночь (2 дня); фуросемид внутрь 0,04 1 раз в день (20 дней); форкан 0,15 внутрь однократно; по 0,05 2 раза в день (7 дней); цифран внутрь 0,5 1 раз в сутки (7 дней); радедорм внутрь 0,005 1 раз в сутки (8 дней); ПУВА-терапия 7 сеансов с
3 капсулами оксаролена; селективная фототерапия 7 сеансов; гелий-неоновый лазер на ладони и подошвы 16 сеансов. Наружно: крем Унны, стероидные средства.
В процессе терапии отмечено лишь незначительное клиническое улучшение и через 2 мес 31.03.03 пациент был повторно госпитализирован в ГУ ЦНИКВИ, где в течение 3 мес получал следующее лечение: ПУВА-терапия с 3 таблетками аммифурина и неотигазона (Ре-ПУВА-терапия); гелий-неоновый лазер на кисти (7 процедур) и феназепам внутрь 0,01 на ночь; супрастин внутрь 0,025 2 раза в сутки; гемодез 400,0 внутривенно капельно (8 вливаний); амитриптилин 0,025 2 раза в сутки; тавегил 0,001 внутрь 2—3 раза в сутки; неотигазон с 28.05.03 внутрь 0,025, затем 0,01 в сутки, далее 0,01 через день; цифран внутрь 0,5 2 раза в день (8 дней). Наружно: крем Унны, аэрозоль СКИН-КАП, мазь целестодерм с гарамицином, 2% салициловая мазь, 10% ихтиоловая мазь, 10% метилурациловая мазь, радевит. Отмечено незначительное улучшение. Больной выписан с рекомендациями: теплые ванны с травами; ожиряющие и взбалтываемые мази (радевит, крем Унны); дипроспан по 1,0 внутримышечно
1 раз в 3 нед. Пациент взят на диспансерный учет по месту жительства, в течение последующих 3 лет изменений клинической картины не отмечалось.
Настоящее наблюдение демонстрирует первично развившуюся и длительно протекающую (4 года) эритродермическую форму болезни Девержи, сопровождающуюся своеобразной клинической картиной: деформацией концевых суставов указательных пальцев, отсутствием участков здоровой кожи на фоне эритродермии; резистентностью к проводимой терапии. Диагноз был определен лишь на основании повторных патогистологических исследований.
Распространенное заболевание среди подростков и молодых людей.По данным нескольких источников волосяной лишай встречается среди 50-80% детей и подростков и почти у 40% взрослых.Появляется в раннем детском возрасте, пик клинических проявлений отмечается в период полового созревания. Женщины страдают несколько чаще, чем мужчины.Отмечается ухудшение состяния в сухую погоду и в зимнии месяцы.Различают:
- идиопатический волосяной лишай;
- наследственную форму (аутосомно-доминантный тип с вариабельной пенетрантностью гена, сообщается о частичной моносомии в коротком плече хромосомы 18 );
- три клинических варианта наследственной формы;
- кератоз фолликулярный шиповидный декальвирующий Сименса;
- надбровную ульэритему;
- червеобразную атрофодермию;
Этиология и патогенез до сих пор полностью не ясны.В результате каких-либо причин образуются пробки из-за избытка кератина, возможно, из-за дефекта адгезии корнеоцитов, в фолликулярном отверстии, что препятствует появлению новых волос. Волосы могут врастать и приводить к перифолликуллярному воспалительному ответу.
![]()
В результате кожа в очагах поражения часто приобретает вид гусиной (цыплячьей), на ощупь она шероховата, грубая, как наждачная бумага.Иногда заболевание бывает генерализованным и поражение охватывает туловище, разгибательные поверхности рук и ног.
Многие авторы выделяют клинические варианты волосяного лишая в отдельные нозологические формы.
Кератоз фолликулярный шиповидный декальвирующий Сименса
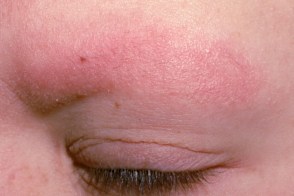
Включает патологию глаз, атрофический фолликулярный кератоз, кератодермию и ониходистрофию. Надбровная ульэритема
![]()
Характеризующееся наличием эритемы и фолликулярного гиперкератоза с преимущественной локализацией в области бровей с дальнейшим развитием поверхностных рубчиков и выпадением волос.Червеобразная атрофодермия
Отличается поражением преимущественно кожи щек. На щеках на фоне легкой эритемы формируются фолликулярные роговые узелки, оставляющие после себя сетевидную атрофию кожи. - Атопический дерматит
- Ихтиоз обыкновенный
- Эритромеланоз фолликулярный лица (эритема, гиперпигментация и волосяной кератоз в области лица),
- Синдром Грэма -Пиккарди-Литтла-Лассюэра (рубцовая алопеция, потеря лобковых и подмышечных волос и волосяной кератоз)
- Сердечно-кожно-лицевой синдром
- Синдром Нунан
- Диабет
- Синдром Дауна
- Гипертрихоз с шерстяными волосами
- Ожирение
- Олмстеда синдром
- Дефицит пролидазы
- Почечная недостаточность
- Гипервитаминоз А
- Монилетрикс
- Пахионихия врожденная
- Эктодермальная дисплазия
- Прием лекарств:системных кортикостероидов, солей лития, биологических препаратов
Диагноз ставиться на основании клинической картины и анамнеза.В редких случаях требуется биопсия, при исследовании гистологического препарата обнаруживается гиперкератоз в виде роговых пробок в устьях волосяных фолликулов, иногда - небольшой перифолликулярный инфильтрат из лимфоцитов.
У многих пациентов состояние улучшается с возрастом, но любая возрастная группа может страдать с детства до старости.Многие пациенты обращаются за лечением, как правило, по косметическим причинам, поскольку очаги пилярного кератоза чаще всего бессимптомные.Расчесы, ношение тесной одежды и применение для самолечения абразивных средств для мытья и скрабов может ухудшить состояние.Эффективного лечения в настоящее время нет.
Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions.
Лечение рефрактерного красного волосяного отрубевидного лишая: проблемы и решения
Gaia Moretta, Erika V De Luca, and Alessandro Di Stefani, Институт дерматологии, Университетская поликлиника Фонда А. Джемелли, Католический Университет Святого Сердца, Рим, Италия
Введение
Красный отрубевидный волосяной лишай (PRP) - редкое хроническое папулосквамозное заболевание с нарушением ороговения, частота которого составляет от 1 на 5000 до 1 на 50000 без гендерной предрасположенности (1).
Патогенез до сих пор неясен: была выдвинута гипотеза, что он вызван аномальным иммунным ответом на различные антигенные стимулы, такие как инфекции, травмы и злокачественные новообразования (2, 3).
Большинство случаев являются спорадическими, но были описаны семейные формы заболевания, в частности, связанные с мутациями в гене CARD . При PRP эпидермис находится в гиперкинетическом состоянии с увеличением пролиферации фолликулярных кератиноцитов.
Было высказано предположение о патогенетической роли дефицита или нарушения функции витамина А (7) или снижения сывороточного уровня ретинолсвязывающего белка, который является носителем витамина А (8), наряду с некоторыми клиническими сходствами с фринодермией (кожным проявлением дефицита витамина А) (9).
Клиническая картина
Как правило, PRP характеризуется небольшими фолликулярными папулами диаметром ~1 мм с центральной кератотической пробкой, сливающимися чешуйчатыми желто-розовыми пятнами и пальмоплантарной кератодермией.
Поражения симметричны и диффузны и появляются сначала на разгибательных поверхностях конечностей, плеч и ягодиц, обычно распространяясь каудально с возможным развитием эритродермии (1, 7).
Дифференциальный диагноз с псориатической эритродермией проводится при определении участков непоражённой кожи. Шелушение встречается часто и по характеру оно мелкопластинчатое, отрубевидное на лице и коже головы и крупнопластинчатое в нижней части тела.
Кожа на ладонно-подошвенных областях имеет тенденцию к утолщению и появлению желто-оранжевого цвета с четко очерченными границами. Иногда пациенты жалуются на лихорадку, озноб, зуд и недомогание (1, 7).
Поражение ногтей также очень часто сопровождается подногтевым гиперкератозом и желто-коричневыми пятнами (10).
Слизистая оболочка полости рта также может быть покрыта белыми пятнами и линиями: эритематозные болезненные высыпания с белыми полосами могут появляться на слизистой оболочке щек, десен и языка, имитируя признаки красного плоского лишая (11).
У взрослых поражения кожи появляются сначала на лице и волосистой части головы, распространяясь в каудальном направлении, в то время как у детей PRP обычно появляется в нижней части тела. Однако клиническая картина и эволюция PRP очень изменчивы.
Классификация
В 1980 году Гриффитс (13-15) классифицировал PRP на следующие пять типов на основе клинических особенностей, возраста начала и прогноза: классический взрослый тип I, атипичный взрослый тип II, классический ювенильный тип III, ограниченный ювенильный тип IV и атипичный ювенильный тип V.
Позже был добавлен VI тип, связанный с вирусом иммунодефицита человека (ВИЧ) (16). Классический взрослый тип I является наиболее распространенным с острым началом и составляет ~50% пациентов. Обычно это начинается с появления одного эритематозного пятна на верхней половине туловища. Последовательно кожные поражения распространяются каудально в течение нескольких недель или месяцев и могут развиться в эритродермию с типичными островками пальмоплантарного желто-оранжевого гиперкератоза. Продолжительность течения этой формы составляет ~3-4 года (13-15). Атипичный взрослый тип II, развившийся у 5% пациентов, имеет хроническое течение до 20 лет. Он характеризуется ихтиоидными поражениями, особенно на коже нижних конечностей, в сочетании с алопецией и участками экзематизации (13-15). Классический ювенильный тип III аналогичен взрослому типу I, но поражает детей (10% пациентов) и имеет более благоприятное клиническое течение, чем у взрослых, обычно наступает ремиссия через 1 год (12). Описанный ювенильный тип IV поражает детей, а также молодых взрослых (25% пациентов); развивается фолликулярный гиперкератоз и эритема, обычно только на коленях и локтях с четко очерченными границами. Пальмоплантарное поражение также характерно при этой форме, а также дорсальное поражение кистей и стоп. В большинстве случаев она остается локализованной, но может характеризоваться ремиссиями и обострениями (12, 17, 18). Атипичный ювенильный тип V встречается в первые несколько лет жизни у 5% пациентов; он хронический и характеризуется фолликулярным гиперкератозом и склеродермическими поражениями кожи верхних и нижних конечностей, в то время как эритема не выражена (13-15).
Связанная с ВИЧ форма VI похожа на тип I, с симметричной зудящей сыпью, представленной эритематозными и шелушащимися фолликулярными папулами, но она имеет более тяжелое течение и, как правило, невосприимчива к лечению.
Заметное закупоривание фолликулов является еще одной распространенной находкой при этой форме; однако другие фолликулярные проявления могут быть связаны с PRP, связанным с ВИЧ, такими как конглобатные угри, гнойный гидраденит и остистый лишай (16).
Эта классификация условная: на самом деле не всегда возможно отнести каждую форму PRP к одному типу, поскольку могут возникать промежуточные формы и переход от одного типа к другому (15).
Диагноз
Диагноз в основном клинический, основанный на вышеуказанных критериях. Однако иногда, особенно когда PRP находится в форме эритродермии, довольно трудно установить четкий клинический диагноз, и его можно ошибочно диагностировать как псориаз, фолликулярную экзему, фолликулярный ихтиоз, генерализованную реакцию гиперчувствительности, Т-клеточную лимфому и lichen planopilaris (1-7).
В последние несколько лет дермоскопия применялась и для оценки воспалительных кожных заболеваний, и она была предложена в качестве возможного дополнительного способа в клинической диагностике, позволяющей лучше дифференцировать PRP от псориаза: псориатические поражения обычно представлены равномерно распределенными точечными сосудами на светло-красном фоне, в то время как поражения при PRP представлены линейными и точечными сосудами и круглыми/овальными желтоватыми областями (19-21).
Недавно два случая PRP у взрослого и подростка были изучены также с помощью конфокальной микроскопии, которая может представлять собой дополнительный инструмент для повышения достоверности диагностики (21).
На самом деле, клинико-патологическая корреляция остается “золотым стандартом” для диагностики PRP, исключая другие дерматозы, особенно псориаз и лимфому кожи.
Типичными гистопатологическими признаками PRP, хотя и не патогномоничными, являются чередующиеся ортокератоз и паракератоз как в вертикальном, так и в горизонтальном направлениях, образующие шахматный рисунок; другими частыми находками являются гипергранулез, закупорка фолликулов, широкие сетчатые гребни, узкие дермальные сосочки и редкий поверхностный периваскулярный лимфоцитарный инфильтрат (22).
Терапия
Лечение PRP включает в себя местную и системную терапию в зависимости от распространенности и тяжести заболевания (таблица 1).
Местное лечение
Местное лечение показано при локализованных формах, таких как PRP III типа, и всегда рекомендуется также в сочетании с системной терапией при тяжелых формах для уменьшения кожных симптомов, таких как зуд и жжение (1, 23, 24).
При локализованных формах PRP основным терапевтическим выбором (23, 24) является местная терапия, включающая кортикостероиды средней и высокой активности, кератолитики, смягчающие средства и производные витамина D, такие как кальципотриол (25), третиноин и тазаротен (18, 26).
Местное лечение в сочетании с системной терапией полезно для лечения гиперкератоза и пальмоплантарной кератодермии, а также для улучшения текстуры кожи, уменьшения шелушения, эритемы и инфильтрации, в частности, рекомендовано применение кератолитиков (5-10% мочевины или 5% салициловой кислоты) и смягчающих средств (27).
Системные методы лечения
Системные методы лечения показаны при заболеваниях средней и тяжелой степени тяжести. Традиционная системная терапия основана в основном на применении ретиноидов, которые фактически рассматриваются как терапия первой линии как у взрослых, так и у детей (1, 27).
К сожалению, существуют рефрактерные случаи, которые не поддаются лечению или рецидивируют после прекращения приема препарата. В этих случаях лечение является сложным, и стандартного терапевтического протокола пока не существует.
Лечение в основном основано на отчетах о случаях заболевания или серии клинических случаев, поскольку из-за редкости заболевания никогда не проводилось контролируемых рандомизированных исследований (27).
Ретиноиды
Системные ретиноиды рассматриваются в качестве системной терапии первой линии для взрослых и детей с PRP, поскольку большинство отчетов о случаях и серий случаев демонстрируют их эффективность (27-39).
Клинический ответ на ретиноиды, как правило, проявляется через 3-6 месяцев терапии, но у некоторых пациентов требуется более длительное лечение. Наиболее часто используются изотретиноин и ацитретин, но есть некоторые данные также об эффективности алитретиноина для PRP (27-30, 35-38).
В ретроспективном исследовании Eastham и соавт. (27) все 12 пациентов, получавших ацитретин, восемь пациентов, получавших ацитретин в монотерапии в дозе 25-50 мг/сут, два пациента, получавшие ацитретин в сочетании с метотрексатом, и два пациента, получавшие ацитретин в комбинации с циклоспорином, сообщили о частичном или значительном улучшении во время лечения.
В проспективном исследовании изотретиноин (2 мг/кг/сут) применялся у 45 пациентов с PRP , и после 4 недель терапии у 28 (62%) пациентов наблюдалось значительное улучшение (29).
В другом ретроспективном исследовании, включавшем 15 пациентов, получавших изотретиноин (в основном 40 мг два раза в день), у 10 (67%) пациентов был полный клиренс в среднем в течение 25 недель (16-44 недель), и только у трех пациентов наблюдалась рефрактерность к лечению (30).
В клинической практике рекомендуемая дозировка изотретиноина для взрослых с PRP составляет 1 мг/кг/сут, в то время как ацитретин используется в дозировке 0,5–0,75 мг/кг/сут. Однако высокие дозы ретиноидов плохо переносятся из-за частых кожно-слизистых побочных эффектов.
Наиболее частыми побочными эффектами ретиноидов являются сухость кожи и слизистых оболочек, гиперлипидемия, повышение уровня трансаминаз и нарушение зрения или оссификации.
У детей ретиноиды могут вызывать гиперостоз и преждевременное закрытие эпифиза, особенно детей в препубертатном возрасте, получавших высокие дозы. Кроме того, ретиноидов следует избегать женщинам детородного возраста из-за их тератогенного эффекта: следует избегать беременности во время и в течение 3 лет после лечения ацитретином и во время и в течение 6 недель после терапии изотретиноином.
Из-за редкости заболевания и отсутствия стандартизированных исследований отсутствуют согласованные данные о продолжительности лечения, риске рецидива при прекращении лечения или необходимости в поддерживающем лечении.
Фототерапия
Доказано, что ультрафиолетовая фототерапия B (UVB) и псорален-ультрафиолетовая терапия A (PUVA) являются успешным способом лечением у некоторых пациентов, страдающих PRP (39, 40), хотя реакция на свет довольно вариабельна. И наоборот, у некоторых пациентов были описаны обострения PRP (8, 41, 42).
Сообщалось, что ацитретин используется в качестве комбинированной терапии с узкополосным УФ-излучением (43), ультрафиолетовым излучением A1 (UVA1) (44) и PUVA (28).
В недавнем отчете о случае PRP у детей было доказано, что фототерапия узкополосным УФ-излучением была успешной после 2 месяцев лечения в сочетании с местными смягчающими средствами, кремом с гидрокортизоном и кальципотриеном, достигая >90% клиренса (45).
Метотрексат
Метотрексат является альтернативным вариантом лечения PRP в качестве средства второй линии (27, 34). В ретроспективных исследованиях была отмечена эффективность еженедельных доз 5-25 мг. Другое ретроспективное исследование продемонстрировало благоприятный ответ у 8/14 пациентов с PRP I типа: средняя продолжительность лечения составила 6 месяцев (от 1 до 12 месяцев) (30).
Другое ретроспективное исследование продемонстрировало эффективность метотрексата в сочетании с ретиноидом, несмотря на повышенный риск гепатотоксичности (33). В другом исследовании, включавшем пять пациентов, было доказано, что метотрексат, рефрактерный к ретиноидам, эффективен (34).
Желудочно-кишечные расстройства является частым побочным эффектом. Дополнительными серьезными побочными эффектами, которые всегда следует проверять, являются панцитопения, гепатотоксичность и пневмонит. Кроме того, его следует избегать женщинам детородного возраста из-за его тератогенного эффекта.
Другие иммунодепрессанты
В частности, циклоспорин можно считать приемлемой терапией для пациентов, которые не реагируют на ретиноиды или метотрексат, и может использоваться как у взрослых, так и при ювенильной формах. Кроме того, эфиры фумаровой кислоты могут быть альтернативным вариантом (51).
Биологические препараты
Учитывая клиническое и гистологическое сходство между псориазом и PRP, все биологические препараты, одобренные для лечения псориаза, также использовались при лечении рефрактерных форм PRP или у пациентов, резистентных или имеющих противопоказания при стандартных системных методах лечения (27, 34).
Поскольку патогенез PRP все еще не ясен, было выдвинуто предположение, что TNF-альфа является ключевым цитокином (52).
Совсем недавно ингибиторы пути IL-23/IL-17 также успешно применялись при PRP, и у пациентов с PRP были обнаружены повышенные уровни IL-17, что дает обоснование для применения этих новых методов лечения (53).
Однако клинический опыт применения биологических препаратов при лечении PRP ограничен отчетами о случаях, а серии случаев и многоцентровые рандомизированные клинические испытания не проводились из-за низкой частоты PRP.
Напротив, также наблюдаются случаи PRP, невосприимчивых к биологическим препаратам, хотя о них редко сообщается, как например, недавно описано в случае PRP IV типа, резистентного к ингибиторам анти-ФНО-альфа и ИЛ-23 (54).
Анти-ФНО-альфа (инфликсимаб, этанерцепт и адалимумаб)
За последние 10 лет ретроспективные исследования и отчеты о случаях показали, что анти-ФНО-альфа-препараты эффективны при лечении PRP как у взрослых, так и у детей (53-64).
Систематический обзор PRP I типа, включающий 15 пациентов, получавших инфликсимаб, этанерцепт или адалимумаб, показал полный ответ у 12/15 пациентов и частичный ответ у 2/15 пациентов. Только в одном случае улучшения не наблюдалось. Шесть пациентов получали анти-ФНО-альфа в качестве монотерапии, в то время как девять пациентов получали лечение в сочетании с ацитретином или метотрексатом (63).
В большинстве случаев ингибиторы ФНО-альфа использовались в качестве средств второй линии при рефрактерном PRP к традиционному системному лечению. В серии случаев из семи пациентов с рефрактерным началом у взрослых PRP доказал эффективность инфликсимаба (три пациента) и этанерцепта (пять пациентов), 5/7 пациентов получали комбинированную терапию низкими дозами ацитретина (0,2 мг/кг/сут).
У всех пациентов наблюдался значительный клиренс на 12-й неделе, и инфликсимаб был ассоциирован с более быстрым ответом, чем этанерцепт. Во время наблюдения только у пациента, страдающего PRP II типа, развился рецидив через 2 месяца после прекращения лечения (51).
В недавнем ретроспективном исследовании 40 пациентов с PRP, проведенном в центре третьего уровня, девять пациентов получали анти-ФНО-альфа-препараты с благоприятным ответом в течение 5-7 недель.
Несмотря на это, большинство из них получали начальную терапию другим системным препаратом (метотрексат, ацитретин или преднизон) (27).
У пациента, страдающего PRP I типа, получавшего адалимумаб и достигшего клинической ремиссии через 4 месяца, были обнаружены повышенные уровни мРНК TNF-альфа в пораженной и окружающей нормальной коже.
Этот вывод согласуется с наблюдаемой клинической ремиссией и поддерживает использование анти-ФНО-альфа для лечения PRP (64).
Ингибиторы IL-23 и IL-17 (устекинумаб и секукинумаб)
В нескольких отчетах о случаях описано применение устекинумаба при PRP I типа, рефрактерном к традиционной системной терапии или анти-ФНО-альфа-агентам (65-69).
Устекинумаб продемонстрировал выраженную и быструю эффективность в уменьшении признаков и симптомов заболевания, а также в долгосрочном контроле заболевания (69).
В недавней серии случаев у трех пациентов, страдающих рефрактерным PRP, был взят образец биопсии пораженной кожи, и была изучена экспрессия мРНК клеток, вырабатывающих врожденные провоспалительные и Т-клеточные цитокины.
Анализ экспрессии генов выявил увеличение цитокинов Т-хелперов (Th) 1 и, в частности, цитокинов Th17, таких как IL-17A, IL-22 и IL-23. У одного пациента уровни IL-17A в образцах пораженной кожи были измерены до и после лечения устекинумабом и снижены после улучшения состояния кожи.
Этот отчет о случае свидетельствует о роли оси IL-23/Th17 в PRP, предоставляя обоснование для таргетной терапии, нацеленной на этот путь в качестве варианта лечения рефрактерного PRP (53). Недавно было показано, что секукинумаб эффективен для лечения PRP в двух отчетах о случаях (70, 71).
В первом случае он был использован у пациента с PRP I типа, рефрактерным к ацитретину (70).
Во втором случае было предложено в качестве приемлемого варианта лечения рефрактерного PRP II типа, при этом улучшение эритемы и пальмоплантарной кератодермии наблюдалось через 2 недели лечения и не было рецидива после 6-месячного наблюдения (71).
Другие методы лечения
Недавний случай доказал эффективность апремиласта в лечении рефрактерного PRP: после 4 недель терапии пациент сообщил о значительном улучшении, а через 6 месяцев наблюдения у него/нее наблюдалось полное исчезновение кожных поражений (72).
Экстракорпоральная фотохимиотерапия также использовалась для лечения двух пациентов с эритродермическим PRP I типа в сочетании с системными ретиноидами и циклоспорином, с хорошими результатами (73). В другом отчете сообщалось, что случай PRP у взрослых II типа был успешно пролечен внутривенным иммуноглобулином (74).
Вывод
PRP-редкое воспалительное заболевание кожи, которое может оказать серьезное влияние на качество жизни пациентов, особенно в хронической и рефрактерной формах. Было испытано много терапевтических вариантов, но у нас нет стандартного протокола для лечения PRP.
Лечение рефрактерных форм остается сложной задачей. Биологические агенты, по-видимому, представляют собой новое эффективное лечение PRP, хотя следует провести многоцентровые рандомизированные клинические испытания, прежде чем рассматривать их в качестве варианта терапии первой линии.
Дальнейшее понимание патогенеза заболевания, возможно, позволит выявить некоторые клинические и фенотипические особенности различных форм PRP в качестве прогностических факторов, позволяющих сделать индивидуальный терапевтический выбор.
Читайте также: