
Системная красная волчанка и эпилептические припадки
Обновлено: 25.04.2024
Ревматические болезни относятся к группе заболеваний, которые характеризуются развитием аутоиммунных процессов против антигенов почти всех органов и тканей организма, что сочетается с образованием аутоантител с органонеспецифическими с
Ревматические болезни относятся к группе заболеваний, которые характеризуются развитием аутоиммунных процессов против антигенов почти всех органов и тканей организма, что сочетается с образованием аутоантител с органонеспецифическими свойствами.
Аутоиммунные процессы осуществляют информационный обмен между нейроэндокринной и иммунной системами, при этом главную роль играют аутоантитела к гормонам, медиаторам и их рецепторам. Продемонстрирован синтез нейропептидов в иммунокомпетентных клетках, а в клетках нейроэндокринной системы доказана возможность синтеза лимфокинов и монокинов.
Функционирование клеток и сигнальная информация обеспечиваются медиаторами и нейротрансмиттерами в обеих системах, между нервной и иммунной системой происходит взаимообмен информацией с помощью цитокинов, стероидов и нейропептидов [1, 2].
Таким образом, доказана общность и взаимосвязь нервной и иммунной систем, сходство между их структурами и функциями и развитие нового направления современной иммунологии — нейроиммунологии [3, 4]. Широкий диапазон неврологических синдромов при аутоиммунных системных заболеваниях позволяет рассматривать их как модельные системы для изучения патогенетической роли иммунных механизмов поражения центральной и периферической нервной системы [5].
Потенциальными мишенями для аутоиммунной агрессии могут быть различные антигены нервной ткани, включая миелин, в том числе ассоциированный с гликопротеином, и его основной белок, ганглиозиды, белок ядер нейрональных клеток и другие [6]. Так, мишеневидные антигены при нейролюпусе представлены антигенами нейрональной ткани, рибосомальным Р-белком, рДНК, малым ядерным рибонуклеопротеидом, а также анионными фосфолипидами при антифосфолипидном синдроме, что обуславливает широкий спектр неврологической симптоматики при данной патологии [7, 8].
По данным различных авторов, частота поражений нервной системы при ревматических заболеваниях (РЗ) колеблется от 40% до 70% и выше, если учитывать психические синдромы и головную боль. Неврологические синдромы включены в классификационные критерии системных васкулитов, опубликованные Американской коллегией ревматологии в 1990 году, в диагностические критерии и критерии активности системной красной волчанки (СКВ), а также в ряд других диагностических критериев, в частности узелкового полиартериита у детей. Неврологические нарушения при РЗ требуют проведения дифференциальной диагностики и назначения адекватного лечения совместно ревматологом и неврологом.
При СКВ в диагностические критерии неврологических поражений включены судороги или психозы. Поражение ЦНС обусловлено в основном сосудистой патологией, к которой относят васкулопатию, тромбозы, истинные васкулиты, инфаркты и геморрагии [7]. В цереброспинальной жидкости обнаруживаются антинейрональные антитела, определяется повышение уровня белка, увеличение клеточного состава. Описаны разные виды судорожных припадков: большие, малые, по типу височной эпилепсии, а также гиперкинезы. При ЦНС-люпусе имеет место головная боль типа мигрени, устойчивая к анальгетикам, но отвечающая на лечение глюкокортикостероидами. Параличи черепных нервов обычно сопровождаются офтальмоплегией, мозжечковыми и пирамидными симптомами и нистагмом. Имеют место зрительные нарушения, преходящие нарушения мозгового кровообращения. Острый поперечный миелит встречается редко и имеет неблагоприятный прогноз. Психические синдромы разнообразны и характеризуются аффективными, органическими мозговыми или шизофреноподобными проявлениями [9, 10].
В рамках СКВ был описан и антифосфолипидный синдром. Этот синдром включает: рецидивирующие артериальные или венозные тромбозы, привычное невынашивание беременности и тромбоцитопению и дополнительные признаки: сетчатое ливедо, неврологические проявления: хорею, эпилепсию, мигренеподобную головную боль, нарушения мозгового кровообращения и деменцию вследствие множественных инфарктов, хронические язвы голеней, Кумбс-положительную гемолитическую анемию, клапанные поражения сердца и серологические маркеры — антифосфолипидные антитела, к которым относятся антикардиолипиновые антитела IgG и IgM и волчаночный антикоагулянт [11].
При системной склеродермии (ССД) неврологический синдром представлен в основном полиневритическими проявлениями, связанными с сосудистыми изменениями и фиброзными процессами в соединительной ткани. Для узелкового полиартериита характерны множественные мононевриты, для гранулематоза Вегенера — асимметричная полинейропатия, для неспецифического аортоартериита — дисциркуляторная энцефалопатия и нарушения мозгового кровообращения.
Собственные данные включали обследование 229 больных различными формами РЗ, среди которых 110 больных страдали системными заболеваниями соединительной ткани: 88 больных СКВ, 22 — ССД и 119 больных — системными васкулитами: облитерирующий тромбангиит (ОТ) — 21, узелковый полиартериит (УП) — 27, неспецифический аортоартериит — (НАА) — 32, геморрагический васкулит (ГВ) — 15 и гранулематоз Вегенера (ГрВ) — 2, другие формы — 22. Проведено детальное неврологическое исследование, ультразвуковая транскраниальная допплерография сосудов мозга, реоэнцефалография, компьютерная (КТ) и магнитно-резонансная томография (МРТ) головного мозга, электроэнцефалография, исследование иммунного статуса.
У большинства больных заболевания дебютировали кожным (50,6%), суставно-мышечным (35,4%) и сосудистым (27,1%) синдромом. Органные поражения в дебюте регистрировались с частотой 7%, синдром артериальной гипертензии — у 5,2%, лихорадка — у 7,0%, гематологические нарушения — 7,9%. Неврологические расстройства в дебюте заболевания отмечены у 12,2% и проявлялись моно- и полиневропатией и синдромом энцефаломиелополирадикулоневрита (ЭМПРН). Поражение периферической нервной системы в дебюте заболевания было особенно характерно для УП и наблюдалось у 30% больных. Основными синдромами дебюта со стороны ЦНС был цефалгический (10,5%) и вестибулярный (6,3%), чаще они наблюдались при НАА. Вовлечение ЦНС имело место у 96 (41,9%) больных, являясь наиболее выраженным при СКВ, НАА, УП.
Цереброваскулярная патология была доминирующей в клинической картине болезни у 34,7% больных, а иногда разнообразные симптомы поражения ЦНС развивались задолго до появления полисиндромной картины заболевания. Основные клинические проявления цереброваскулярной патологии включали: цефалгический (82%), астенический (76%), вестибулярно-атактический (80%), пирамидный (74%) синдромы, синдром вегетативно-сосудистой недостаточности (69%), диссомнический (79%) и базально-оболочечный (37%), гипопоталамической дисфункции (34,7%).
Описанная неврологическая симптоматика часто сочеталась с симптомами сосудистой недостаточности головного мозга, которые объединялись синдромом дисциркуляторной энцефалопатии 1 (11%), 2 (26,4%) или 3 (8%) степени. У 7,8% больных имели место преходящие нарушения мозгового кровообращения.
Гипоталамическая дисфункция у больных РЗ проявлялась полиморфными нейроэндокринными расстройствами, нарушением терморегуляции, преимущественно по типу пароксизмальной центральной гипертермии, инсомнией, патологией психоэмоциональной сферы.
Установлено достоверное преобладание у больных пирамидной недостаточности слева (41%). Преобладание пирамидной недостаточности справа регистрировалось реже (23,7%). Дистонические феномены в форме вестибулярно-мозжечковой установки кисти и диссоциированная мышечная гипотония в ногах также были более выражены слева. Полученные данные свидетельствуют о преобладающем поражении пирамидной и сенсорной систем, а также неспецифических структур правого полушария, которое тесно связано с гипоталамической областью и обеспечивает адаптацию организма к воздействующим факторам внешней среды. Обнаруженная функциональная асимметрия свидетельствует о срыве адаптационных механизмов нервной системы и указывает на роль дисфункции правополушарно-гипоталамической системы.
При использовании методов МРТ и/или КТ было выявлено изменение желудочковой системы в виде ее расширения или деформации и/или расширения субарахноидального пространства, а также очаговые поражения различных структур головного мозга, атрофия вещества мозга и краниовертебральные аномалии. Признаки наружной, внутренней или сочетанной гидроцефалии отмечались при всех нозологических формах. Очаговые изменения вещества мозга включали гиперденситивные зоны, гиподенситивные зоны с отеком или без него, единичные или множественные.
При исследовании сосудистой системы и мозгового кровообращения достоверно наблюдалось повышение тонуса сосудов, гипертонический и дисциркуляторный тип кровообращения по данным реоэнцефалографии (РЭГ) и увеличение линейной скорости кровотока по средней мозговой артерии. Больные с вовлечением ЦНС отличались по электроэнцефалографии: им были свойственны диффузные патологические изменения, наличие дезорганизации альфа-ритма, дизритмий и пароксизмальной активности.
Корреляционный анализ цереброваскулярной патологии и результатов инструментальных исследований сосудов головного мозга показал, что при всех нозологических формах у пациентов имело место нарушение венозной гемоциркуляции. В последующем происходило сужение церебральных артерий, ликвородинамические нарушения с формированием внутричерепной гипертензии, нарушением системы микроциркуляции в головном мозге. Очаговые поражения головного мозга отличались локализацией процесса в зависимости от нозологической формы. В табл. представлены основные неврологические проявления РЗ.
Среди больных РЗ цереброваскулиты (ЦВ) имели место у 28,3% больных. Диагноз ЦВ ставился при обнаружении очаговой неврологической симптоматики, изменений на глазном дне, снижения зрения, наличии признаков нарушения мозгового кровообращения, а также по результатам КТ и ядерно-магнитно-резонансной томографии (ЯМРТ), при которых выявлялись наружная и внутренняя гидроцефалия, очаговые изменения в коре и субкортикальном веществе. При этом с течением времени число очагов любой локализации в головном мозге нарастало. При магнитно-резонансном ангиографическом (МРА) исследовании отмечались множественные сегментарные неровности сосудистой стенки, циркулярные или эксцентричные стенозы и дилятация мелких и средних интракраниальных артерий с формированием аневризм, нарушения кровотока. Выявленное снижение интенсивности МРА-сигнала на фоне повышения активности ревматического процесса свидетельствовало о наличии ЦВ.
Иммунологическими маркерами ЦВ считали антитела к нативной ДНК, IgG-антитела к кардиолипину (аКЛ) и IgM аКЛ, антинейтрофильные цитоплазматические антитела (АНЦА), в меньшей степени — РФ и волчаночный антикоагулянт (ВА). Имели место клинико-иммунологические корреляции с неврологическими проявлениями.
Изолированный (первичный) ЦВ характеризовался обнаружением симптомов вовлечения ЦНС и такими признаками, как головная боль, судороги, менингеальный синдром, острая прогрессирующая энцефалопатия без признаков экстракраниального или системного васкулита, психические синдромы, деменция, прогрессирующее снижение интеллекта, инсульты, нарушения зрения, нистагм. Чаще перивентрикулярные очаги выявлялись в первый год заболевания.
Ряд больных обращались на консультацию к окулисту в связи с ухудшением зрения, вплоть до амавроза, наличием увеита, ишемического неврита. Ангиопатия сетчатки имела место у 41% этих больных, флебопатия — у 14%, ретиноваскулит — у 6%, ангиоспазм — у 13%, ангиосклероз — у 18%.
Полиневритический синдром встречался у подавляющего большинства больных (96,7%) в виде сенсорной, чувствительно-двигательной полиневропатии или в сочетании с поражениями ЦНС, с синдромом ЭПН и ЭМПРН. При ССД, ОТ и ГВ преобладали формы в виде чувствительной или чувствительно-двигательной полиневропатии, а при СКВ и НАА — формы с сочетанным поражением периферической НС (ПНС) и ЦНС — синдромы ЭПН и ЭМПРН. При ОТ и НАА отмечалась отчетливая диссоциация степени выраженности полиневропатии по оси тела, причем при ОТ симптоматика была более отчетливой в ногах, при НАА — в руках. В целом асимметричная полиневропатия имела место у 19,2% больных, достигая максимума при УП (59,3%).
Патология НС при РЗ нередко определяет прогноз, клиническую картину заболевания и качество жизни больных, а также требует обязательного сочетанного применения базисной противовоспалительной терапии, ангио- и нейропротекторов. К группе нейропротекторов относят Актовегин, Инстенон. Используются препараты, улучшающие мозговое кровообращение, — Винпоцетин, Кавинтон, метаболические средства с антигипоксантным действием — Ноотропил, Пирацетам, Церебролизин, по показаниям седативные и противосудорожные средства, антидепрессанты.
При РЗ терапия включает глюкокортикостероиды, иммуносупрессанты, иммуноглобулин, плазмаферез, иммуномодуляторы, дезагреганты, нестероидные противовоспалительные препараты и симптоматические средства.
Лечение состоит из нескольких этапов: быстрое подавление иммунного ответа в дебюте заболевания и при его обострениях (индукция ремиссии); длительная поддерживающая терапия иммуносупрессантами, в дозах, достаточных для достижения клинической и лабораторной ремиссии заболевания; определение степени повреждения органов или систем организма и их коррекция, проведение последующих реабилитационных мероприятий.
Первый этап включает эффективное подавление иммунного воспаления на ранних стадиях болезни и предполагает использование глюкокортикостероидов, иммуносупрессантов цитостатического действия типа Циклофосфана и антиметаболитного действия типа Метотрексата, цитокинсупрессивного препарата Циклоспорина А, внутривенного иммуноглобулина, назначение повторных курсов пульс-терапии метилпреднизолоном и Циклофосфаном, в сочетании с экстракорпоральными методами лечения.
При острых церебральных нарушениях при высокой активности СКВ используется схема пульс-терапии с введением Метипреда 1 г внутривенно 1 раз в день в течение 3 дней и с добавлением 800 мг Циклофосфана во 2-й день. При хроническом течении СКВ суточная доза преднизолона составляла 15–20 мг с последующим постепенным снижением, Циклофосфан применяется внутримышечно в дозе 400 мг в неделю до 1600–2000 мг на курс, затем по 200 мг в неделю в течение года и более. Апробируются препараты мофетила микофенолат и лефлуномид.
При патологии органа зрения назначают нестероидные противовоспалительные препараты в виде инъекций диклофенака, а затем пероральные препараты этой группы, дезагреганты, при наличии признаков воспалительной активности добавляют умеренные дозы глюкокортикостероидов, а при резком снижении зрения и выраженных признаках активности используют пульс-терапию.
Проводится определение наиболее эффективных и менее токсичных схем применения иммуносупрессивных препаратов, путей их введения, и включение в комплексное лечение больных препаратов, улучшающих микроциркуляцию и/или влияющих на реологические свойства крови (Гепарин, Фраксипарин, Трентал, Ралофект, Тиклид).
В ряде случаев назначают препараты типа Реаферона, а при наличии инфицированных язв, некрозов кожи или конечностей применяют антибиотики. Ввиду многообразия нозологических форм на выбор лекарственных средств в дебюте заболевания оказывает влияние распространенность патологического процесса и наличие интеркуррентной инфекции. Показано назначение ангиопротекторов и посиндромная терапия.
Учитывая высокий удельный вес неврологической патологии, больные РЗ должны проходить комплексное клинико-инструментальное неврологическое исследование уже на ранней стадии патологического процесса. Постановка диагноза РЗ и комплексная терапия глюкокортикостероидами и иммуносупрессантами способствуют коррекции нарушений ЦНС и ПНС.
Литература
Stenberg E. M. Neuroendocrine regulation of autoimmune / inflammatory diseases // J. Endocrinol. 2001; 169 (3): 429–435.
Насонова В. А., Иванова М. М., Калашникова Е. А. и др. Актуальные проблемы нейроиммунологии // Вестн. РАМН. 1994;1: 4–7.
Сrofford L. J. The hypothalamic-pituitary-adrenal axis in the pathogenesis of rheumatic diseases // Endocrinol. Metab. Clin. North. Am. 2002; 31 (1): 1–13.
Мотовилов А. А., Травина И. В., Проказова Н. В. и др. Антитела к нейтральным гликосфинголипидам и ганглиозидам у больных системной красной волчанкой с поражением центральной нервной системы // Клин. ревматол. 1995; 1: 36–38.
Иванова М. М. ЦНС-люпус: проблемы и достижения (результаты 10-летнего клинико-инструментального исследования) // Тер. арх. 2001; 5: 25–29.
Лисицина Т. А., Вельтищев Д. Ю., Серавина О. Ф. и др. Варианты психических нарушений у больных системной красной волчанкой // Научно-практ. ревматол. 2008; 4: 21–27.
Nived O., Sturfelt G., Liang M. H., De Pablo P. The ACR nomenclature for CNS lupus revisited // Lupus. 2003; 12: 872–876.
Эпилептические припадки являются одним из самых частых симптомов поражения центральной нервной системы (ЦНС) при системной красной волчанке (СКВ) как у взрослых пациентов, так и у детей. Не случайно они включены в номенклатуру нейропсихических расстройств Американской организации ревматологов [53].
Частота эпилептических припадков при СКВ
Распространенность эпилептических приступов различается в зависимости от возраста больных и этапа развития основного заболевания.

Эпилептические припадки среди детей с СКВ определяются частотой 9,4-84,4%, нередко являясь первым клиническим симптомом этого заболевания [59] (табл. 1).

В ходе ретроспективного изучения 91 ребенка с СКВ M. Steinlin и соавт. [51] выявили, что в 48% случаев СКВ манифестировала неврологическими симптомами и эпилептические припадки, как дебютный симптом, наблюдались у 20% больных. Но они могут возникать и через несколько лет после постановки диагноза СКВ [19, 30, 42, 45]. Крупное сравнительное исследование по эпилепсии среди детской и взрослой популяции больных с СКВ было проведено H. Gomez и соавт. [17], которые, обследовав 870 пациентов, у 98 из них обнаружили неврологическую и психиатрическую патологию. Эпилептические припадки в детской группе (40 больных в возрасте до 16 лет) были одним из самых распространенных неврологических симптомов - в 20%, а среди взрослых (88) они были выявлены в 14% случаев. По данным других авторов [4, 36, 39, 46], у взрослых пациентов с СКВ эпилептические припадки развиваются в 3-42,9% случаев (табл. 2). Как у детей, так и у взрослых пациентов эпилептические припадки могут быть первым манифестным симптомом этого системного ревматического заболевания, возникая до постановки основного диагноза, а также после его установления.
Другое заслуживающее внимание исследование провели R. Andrade и соавт. [4], обследовавшие 600 пациентов с СКВ, из которых у 40 (6,7%) имелись эпилептические припадки. По данным проведенного авторами мультивариационного анализа, активность заболевания и молодой возраст были независимыми факторами риска развития эпилептическими припадков непосредственно после манифестации СКВ (p=0,0004 и p=0,0304 соответственно), в то время как длительное использование гидроксихлорокина оказалось независимым фактором риска возникновения эпиприпадков в последние годы после дебюта ревматического заболевания (p=0,0131).
Патогенез эпилептических припадков
Большинством авторов [10, 11, 15, 23, 24, 27, 31, 38, 41, 48, 49, 55] в проведенных исследованиях анализировались факторы риска, ассоциирующиеся с развитием эпилептических припадков, однако до настоящего времени их патогенез при СКВ остается до конца не изученным. В качестве возможных механизмов рассматриваются перенесенные цереброваскулярные заболевания и влияние антител, повреждающих церебральную ткань [1, 20, 23, 50, 56]. Эпилептические приступы как у взрослых, так и у детей с СКВ наиболее часто связаны с антифосфолипидными [9, 35, 48], а также с антирибосомальными [12], антимозговыми [58] и анти-Ro [22] антителами. J. Mikdashi и соавт. [37] в течение 12 лет наблюдали за 195 пациентами с СКВ, эпилептические припадки были зафиксированы у 28 (14%). Согласно Международной классификации эпилепсий у 21 пациента были генерализованные и у 7 - парциальные эпилептические припадки. Повторные приступы встречались у 12 (43%) из 28 больных. Определенными клиническими признаками, лежащими в основе развития судорог, были активность болезни, наличие умеренных или высоких титров антикардиолипиновых и анти-Smith антител, нарастающее повреждение, предшествующий инсульт и мужской пол.
В ряде исследований [3, 31, 48, 49, 60] была показана ассоциация эпилептических припадков с атрофией коры головного мозга, выявляемой с помощью рентгеновской или магнитно-резонансной компьютерной томографии (КТ, МРТ). Так, V. Zanardi и соавт. [60] при обследовании 107 пациентов с СКВ с различными нейропсихическими синдромами отметили, что самым частым клиническим проявлением среди пациентов с атрофией коры головного мозга по данным нейровизуализации были эпилептические припадки. В то же время F. Joseph и соавт. [25] при ретроспективном анализе 41 пациента с СКВ из Юго-Западной Англии и Южного Уэльса показали отсутствие структурных изменений или наличие неспецифических изменений головного мозга по результатам МРТ у пациентов с эпилепсией.
В литературе упоминаются и другие причины развития эпилептических припадков при СКВ: церебральный васкулит [28], паренхиматозное кровоизлияние [57], микроэмболию [7], длительное лечение ритуксимабом [54], поражение ЦНС в виде Reversible posterior leucoencephalopathy syndrome (RPLS) [44], множественные мелкие инфаркты в нескольких областях головного мозга [22].
ЭЭГ при СКВ с эпилептическими припадками
Эпилептические припадки при СКВ
Что касается эпилептических припадков при СКВ, их клинических проявлений и особенностей, то они, согласно результатам многих зарубежных исследований [5, 8, 11, 31, 33, 34, 37, 50, 51], могут протекать в самых различных формах - генерализованные тонико-клонические, тонические судороги, простые и сложные парциальные приступы, миоклонические эпилептические припадки, рефлекторные приступы и эпилептический статус - как единичные, так и повторные.
Приведенные выше данные зарубежных исследователей согласуются с нашими собственными наблюдениями: среди 139 пациентов эпилептические припадки выявлялись у 9 (6,6%). Эпилептические приступы развивались одинаково часто на различных этапах развития заболевания: до 1 года от постановки диагноза СКВ - у 22,2% больных, от 1 года до 3 лет - у 22,2%, от 3 до 5 лет - у 22,2% и через 10 лет и позднее - у 33,4%. Согласно Международной классификации эпилепсии и эпилептических синдромов (1981) парциальные приступы отмечались в 22,2% наблюдений и генерализованные - в 77,8%. На МРТ головного моза у всех пациентов определялась корковая атрофия, в 33,3% случаев - корковые очаги и в 22,2% - перивентрикулярные очаги. На межприступной ЭЭГ патологические изменения отсутствовали у 22,2% больных, локальная эпилептическая активность регистрировалась у 33,3% и диффузные изменения - у 44,4%. Не получено достоверной зависимости развития приступов от активности основного заболевания.
Таким образом, при СКВ эпилептические приступы могут развиваться в любом возрасте, быть первичными в результате поражения ЦНС на фоне активности свойственного основному заболеванию иммунопатологического процесса, вследствие церебрального васкулита (ишемического или геморрагического поражения), кардиальной эмболии, а также возникать независимо от активности СКВ, определяясь сопутствующими инфекциями, уремией, гипертензией, лекарственной интоксикацией, электролитными или метаболическими сдвигами.
Кафедра неврологии и нейрохирургии Ярославской государственной медицинской академии
Эпилептические приступы у пациентов с системными ревматическими заболеваниями
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2014;114(4‑2): 59‑65
Пизова Н.В. Эпилептические приступы у пациентов с системными ревматическими заболеваниями. Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2014;114(4‑2):59‑65.
Pizova NV. Epileptic seizures in patients with systemic rheumatic disorders. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2014;114(4‑2):59‑65. ().
Кафедра неврологии и нейрохирургии Ярославской государственной медицинской академии
При системных ревматических заболеваниях в болезненный процесс вовлекаются многие органы. Судороги являются одним из наиболее распространенных неврологических проявлений, а иногда могут быть дебютным симптомом. В представленном обзоре литературы рассматриваются механизмы, лежащие в основе эпилептических приступов у пациентов с системными аутоиммунными заболеваниями, и предрасполагающие факторы, в том числе сосудистые заболевания (например, протромботическое состояние, антитела к кардиолипину, эмболии, васкулит), антинейрональные антитела, иммунные комплексы, цитокины, нарушения обмена веществ, инфекции и лекарственные препараты. Обсуждаются вопросы необходимости индивидуализации диагностических и терапевтических стратегий с учетом вида приступов и особенностей пациента.
Кафедра неврологии и нейрохирургии Ярославской государственной медицинской академии
При системных ревматических заболеваниях в патологический процесс вовлекаются многие органы и системы организма, включая центральную (ЦНС) и периферическую нервную систему [1].

Эпилептические приступы в этих случаях являются одними из наиболее распространенных неврологических проявлений (табл. 1).
Причины появления эпилептических приступов у пациентов с системными ревматическими заболеваниями множественны. В некоторых случаях такие приступы являются проявлением метаболических нарушений, инфекционных заболеваний, токсического действия лекарств, в других случаях появление судорог может говорить об опасном для жизни прогрессировании основного заболевания. Ниже приведен перечень соответствующих заболеваний.
Причины эпилептических приступов у пациентов с системными ревматическими заболеваниями:
1. Сосудистые заболевания
3. Антинейрональные антитела
4. Иммунные комплексы
6. Метаболические расстройства
7. Инфекционные заболевания
8. Лекарственные препараты
9. Неуточненные причины.
Существуют и случаи, где причина развития эпилептических приступов остается неуточненной.
Рассмотрим теперь отдельные ревматические заболевания.

Системная красная волчанка. Эпилептические приступы рассматриваются как важное проявление неврологических синдромов при СКВ [2, 3]. По разным данным, они развиваются в 8,3-25% случаев СКВ (табл. 2) 11. Это означает, что их частота почти в 8 раз выше распространенности эпилепсии в общем населении. Так, по данным E. Kampylafka и соавт. [12], которые обследовали 370 пациентов с СКВ (средний возраст 32±14 лет, 88% женщины) и средней длительностью заболевания 9±7,8 года судороги наблюдались в 35% случаев. Все эпилептические приступы протекали по типу тонико-клонических, у 3 пациентов был эпилептический статус.
В другом исследовании [10] на основании осмотра 128 пациентов с СКВ (120 женщин и 8 мужчин) эпилептические приступы были выявлены в 21% случаев (16% - парциальные). Среди 282 китайских пациентов с СКВ (средний возраст 31,8±14 лет) и средней длительностью болезни 6,7 года эпилептические приступы выявлялись в 20% [5].
Эпилептические приступы являются одним из самых частых неврологических проявлений и у детей с СКВ, встречаясь, по разным данным, у 9,4-84,4% пациентов (табл. 2) 14. Так, среди 86 детей с СКВ 16 детей имели судороги, а у 12 судороги отмечались как инициальное проявление поражения ЦНС [17].
H. Liou и соавт. [23], а позднее и другие авторы [24] обнаружили, что эпилепсия в 3,7 раза встречается чаще среди пациентов с СКВ, которые имели антикардиолипиновые антитела (аКЛ), чем у пациентов с СКВ без них. У пациентов с СКВ была также выявлена корреляция между повышенным уровнем антител к субъединице NR2 NMDA-рецепторов в ЦСЖ, но не в сыворотке, и нейропсихическими синдромами, включая судороги [25, 26]. Уровень анти-Р рибосомальных антител повышается при вовлечение ЦНС, в том числе при психозах и судорогах 28. С другой стороны, отмечено, что низкий риск развития судорог коррелирует с высоким уровнем анти-La-антител и применением антималярийных препаратов [30, 31].
Также выявлено [30], что активность заболевания и молодой возраст были независимыми факторами риска развития эпилептических приступов в течение короткого времени от момента клинической манифестации СКВ (р=0,0004 и р=0,0304 соответственно), в то время как наличие поражения кожи и слизистых, а также использование гидроксихлорокина были независимыми факторами риска возникновения приступов через более длительный период от манифестации основного заболевания (р=0,0131 и р=0,0039 соответственно). Связь возникновения эпилептических приступов с высокой оценкой SLEDAI (р=0,07) и более молодым возрастом начала заболевания (р=0,08) была отмечена и другими исследователями [12].
Представляет интерес исследование J. Mikdashi и соавт. [9], которые в течение 12 лет наблюдали 195 пациентов с СКВ. У 28 (14%) из них за этот период были зафиксированы эпилептические приступы. Согласно международной классификации эпилепсии у 21 пациента были генерализованные приступы и у 7 - парциальные. За время наблюдения повторные приступы встречались у 12 из 28 больных СКВ (43%). Развитие эпилептических приступов было связано с активностью болезни, особенно с наличием психозов, умеренными или высокими титрами аКЛ и анти-Smith-антител. Эти авторы также отметили, что сопутствующие неврологические нарушения, предшествующий инсульт и мужской пол были предикторами развития эпилепсии.
Около четверти приступов у больных СКВ являются клинически определенными симптомами в виде парциальных моторных или сенсорных приступов, которые могут быть связаны с локальным патологическим процессом в головном мозге. Наиболее частой причиной эпилепсии у больных СКВ является инсульт. Наиболее распространенными типами приступов в этой группе больных, как правило, являются простые или сложные парциальные приступы с или без вторичной генерализации. Основной причиной очагового поражения мозга или тромбоза является кардиальная эмболия, при этом могут поражаться как крупные сосуды, так и микроциркуляторное русло или может быть первичный васкулит [2].
Судороги могут развиваться не только на фоне активного системного заболевания, но и быть от него относительно изолированными. Так, в одном из исследований [32] эпилептические приступы были связаны с высокой активностью заболевания по ECLAM (р=0,02), его поздним началом (р=0,03), наличием в анамнезе серозитов (р<0,01) и гломерулонефрита (р=0,03); эти наблюдения соответствуют данным о том, что эпилептические приступы чаще встречаются у пациентов с волчаночным нефритом.
Стероидная терапия, особенно высокие дозы пульс-терапии, может вызывать эпилептический статус [33]. Другими возможными причинами приступов служат инфекции, уремия, гипертония. Возникновение эпилептических приступов у пациентов с СКВ может быть проявлением и обратимой задней лейкоэнцефалопатии [34].
Необходимо отметить, что эпилептические приступы у пациентов с СКВ могут быть не связаны с основным аутоиммунным заболеванием, быть проявлением самостоятельного заболевания - эпилепсии. Так, T. Toyota и соавт. [35] среди всех пациентов с СКВ в период с января 2000 по август 2011 г. диагностировали эпилепсию в 17 случаях. 7 пациентов имели медиальную височную эпилепсию, 8 - симптоматическую эпилепсию после инсульта и у 2 была генерализованная эпилепсия. Из 7 пациентов с медиальной височной эпилепсией переднетемпоральные спайки были отмечены у всех больных на ЭЭГ, а на МРТ были изменения, свидетельствующие о наличии склероза гиппокампа у 4 пациентов.
При синдроме Шегрена эпилептические приступы, парциальные или генерализованные, встречаются в 3% случаев [36] и у 8,5% пациентов с неврологическими проявлениями [37].
У пациентов с гранулематозом Вегенера на ранних этапах эпилептические приступы развиваются редко [38], но встречаются примерно у 10% пациентов с неврологическими проявлениями [39]. В то же время в литературе имеются наблюдения, когда ГВ дебютировал с вторично-генерализованных эпилептических приступов у 17-летней женщины с гранулематозными очагами в головном мозге [38]. Эпилептические приступы при рассматриваемом заболевании могут быть тонико-клоническими, парциальными и миоклоническими.
Эпилептические приступы - это одно из проявлений поражения головного мозга у пациентов с неспецифическим аортоартериитом 55. Z. Bolaman и соавт. [58] отметили наличие эпилептических приступов менее чем у 10% больных [58], а по данным Z. Li-xin и соавт. [59], они встречаются у 25,9% пациентов [59]. Эпилептические приступы в качестве случаев манифестного проявления неспецифического аортоартериита отмечались, но редко, и в большинстве случаев они носили повторный характер [60, 61]. Так, Z. Bolaman и соавт. [58] описали 15-летнюю девушку, у которой в течение 15 дней были генерализованные тонико-клонические приступы с потерей сознания, продолжавшиеся в течение 10 мин, артериоаортография показала стенозы подключичной артерии и стенозы почечных артерий, а на МРТ головного мозга выявлялось патологическое изменение сигнала от глубокого белого вещества с двух сторон. Церебральная ангиография выявила локальный стеноз сосудов головного мозга, что было классифицировано как тип V артериита Такаясу в соответствии с классификацией 1994 г. Сходный случай был описан бразильскими исследователями [62] у ребенка 3 лет, которому диагноз основного заболевания был выставлен после возникновения у него тонико-клонического эпилептического статуса.
Как и у пациентов с СКВ, при неспецифическом аортоартериите редко развивается синдром обратимой задней лейкоэнцефалопатии, который характеризуется головной болью, спутанностью сознания, нарушением зрения, эпилетическими приступами и преходящими обратимыми изменениями на МРТ в корковом и подкорковом белом веществе теменно-затылочных долей [63, 64].
Синдром Гудпасчера - редкое заболевание, характеризующееся быстро прогрессирующим гломерулонефритом, диффузными легочными кровотечениями и наличием антител к базальной мембране клубочков почек (анти-БМК). Поражение ЦНС при синдроме Гудпасчера встречаются крайне редко.
В литературе описано всего несколько случаев [65, 66]. У всех пациентов отмечались повторные эпилептические приступы, с или без кровохарканья. Результаты МРТ головного мозга этих пациентов показали диффузные лакунарные инфаркты [65] или множественную корковую ишемию в затылочных и теменных долях [66, 67]. G. Pérez-Suárez и соавт. [68] сообщили о 20-летнем мужчине с синдромом Гудпасчера, клинически у которого заболевание проявлялось кровавой мокротой, тонико-клоническими эпилептическими приступами и высокими титрами антител к базальной мембране клубочков почек (анти-БМК). Еще один случай отражен при описании 34-летнего мужчины [69], у которого развились генерализованные тонико-клонические судороги и правосторонний гемипарез. На МРТ головного мозга на Т2-взвешенных изображениях наблюдались мелкие очаги гиперинтенсивного сигнала с двух сторон в полушариях мозжечка, правой лобной и левой теменной долях. При контрастировании на Т1-взвешенных изображения наблюдалось лептоменингиальное утолщение в обоих парасагиттальных регионах и полушариях мозжечка, что соответствовало множественным небольшим сосудистым инфарктам.
Эпилептические приступы описаны и при пурпуре Шенлейна-Геноха, чаще у детей и реже у взрослых пациентов [70, 71]. Так, у детей с геморрагическим васкулитом неврологические расстройства наблюдаются до 7% случаев [72, 73] и 40% из них проявляются эпилептическими приступами. Генерализованные приступы являются наиболее распространенными [73].
По данным нескольких исследований 74, среди пациентов с геморрагическим васкулитом эпилептические приступы развились в 53% случаев, включая приступы с и без вторичной генерализации, парциальные судороги со сложной симптоматикой [76], генерализованные судороги и эпилептический статус [74, 77]. L. Garzoni и соавт. [78] показали, что среди 54 пациентов с геморрагическим васкулитом, имеющих поражение нервной системы, парциальные эпилептические приступы наблюдались в 7% случаев, генерализованные - в 33%. Более того, преходящие патологические изменения на ЭЭГ, включая фокальную или генерализованную медленноволновую активность, острые волны, а иногда и пароксизмальную активность отмечали в 21 (55%) из 39 случаев.
При проспективном наблюдении 26 детей с пурпурой Шенлейна-Геноха без неврологических расстройств транзиторные патологические фокальные изменения на ЭЭГ были выявлены в 50% случаев [79]. Однако авторы сделали заключение, что эти изменения могут быть нормальными для определенного возраста. Для диагностики церебрального васкулита при пурпуре Шенлейна-Геноха и преходящих ишемических изменений на фоне васкулита может использоваться МРТ 80.
При геморрагическом васкулите судороги могут быть предвестником начала болезни и появляться до других системных проявлений [73, 74] или могут появляться после кожных, ренальных и гастроинтенстинальных симптомов [73, 83]. Эпилептические приступы могут быть связаны с тяжелой гипертензией или метаболическими нарушениям или они могут быть признаком субарахноидального [74, 77], субдурального [77], корковых [77] или интрапаренхиматозных кровоизлияний [84]. Они могут развиваться и в отсутствие этих провоцирующих факторов [73, 74]. В этом отношении возникновение судорог может отражать нейрональную дисфункцию из-за ишемии, вызванной церебральным васкулитом. Прогноз различен. Эпилептические приступы могут прекратиться или рецидивировать, в части случаев становятся резистентными [74, 77].
При узелковом полиартериите парциальные или генерализованные эпилептические приступы встречаются в 4-20% случаев [85]. На ЭЭГ при этом выявляются как неспецифические изменения, так и полное отсутствие патологических изменений [85]. В большинстве случаев возникновение эпилептических приступов связано с активностью основного заболевания, но при отсутствии структурных изменений. Описан случай развития эпилептических приступов при узелковом полиартериите как проявление синдрома обратимой задней лейкоэнцефалопатии у женщины [86].
В литературе встречаются единичные описания развития эпилептических приступов у пациентов с другими системными ревматическими заболеваниями: при дерматозите у 10-летнего ребенка [87], у детей при болезни Кавасаки [88].
Таким образом, эпилептические приступы могут развиваться при различных системных ревматических заболеваниях, наиболее часто при системной красной волчанке, неспецифическом аортоартериите, пурпуре Шенлейна-Геноха и болезни Бахчета. Причины и патогенез эпилептических приступов остаются до конца не известными.
Что такое юношеская миоклоническая эпилепсия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Агранович А. О., эпилептолога со стажем в 12 лет.
Над статьей доктора Агранович А. О. работали литературный редактор Вера Васина , научный редактор Татьяна Гаврилова (Уханова) и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Юношеской миоклонической эпилепсией (синдромом Янца) называют эпилептический синдром, который проявляется внезапными подёргиваниями в мышцах — миоклоническими приступами (от греч. "myos" — мышца, "klonos" — беспорядочное движение). Заболевание обычно развивается в подростковом возрасте.
Подёргивания в первую очередь возникают в мышцах верхнего плечевого пояса и рук. Сначала пациенты не обращают на них внимания, но со временем эпизоды возникают всё чаще и ухудшают качество жизни. Например, во время приступов из рук могут выпадать предметы. В дальнейшем появляются подёргивания ног, из-за которых человек может упасть.
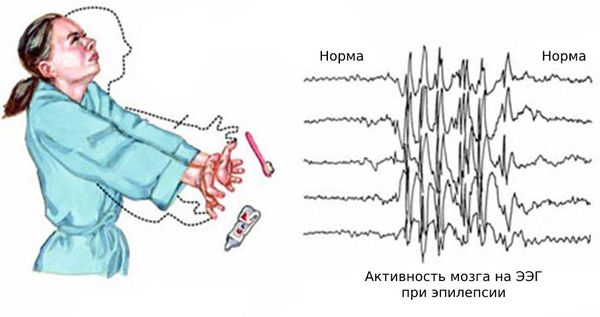
Нередко к этим эпизодам присоединяются генерализованные судорожные приступы — судороги возникают по всему телу и пациент теряет сознание. Также возникают абсансы — бессудорожные приступы с отключением сознания и амнезией на этот период. Как правило, частота генерализованных приступов невысокая: от одного за всю жизнь до раза в месяц. Подёргивания обычно случаются утром после пробуждения. Ярким провоцирующим фактором может стать недосыпание или вынужденное пробуждение. Также в трети случаев отмечается фотосенситивность — чувствительность к ритмическим вспышкам света.
Распространённость
Юношеская миоклоническая эпилепсия составляет 5–10 % среди всех эпилепсий и чуть больше четверти среди генетических генерализованных эпилепсий [5] . Заболевание проявляется в возрасте от 7 до 21 года, чаще в 11–15 лет, и более распространено среди женщин (61 %) [2] .
Причины заболевания
По классификации Международной противоэпилептической лиги за 2017 год, юношеская миоклоническая эпилепсия относится к генетическим болезням [1] . Заболевание имеет полигенное наследование, то есть контролируется двумя или более генами. Его развитие связано с локусами (участками ДНК): 6p11-12 (EJM1), 15q14 (EJM2), 6р21 (EJM3), 5q12-q14 (EJM4), 5q34-q35 (EJM5), 2q22-q23 (EJM6), 1p36 (EJM7), 3q26 (EJM8), 2q33-q36 (EJM9). Выделить ген, сильнее всего влияющий на развитие заболевание, пока не удалось [3] [4] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы юношеской миоклонической эпилепсии
Основным симптомом заболевания являются миоклонические вздрагивания (миоклонии) — патологические непроизвольные сокращения мышц или их групп [7] . Во время приступа пациенты испытывают ощущение, похожее на лёгкий удар током. В ответ на него возникают молниеносные движения в мышцах: от лёгкого подёргивания кончиков пальцев до патологического вздрагивания всего тела, которое может привести к падению.
Чаще всего подёргивания возникают в верхнем плечевом поясе: мышцах рук и плеч с обоих сторон. Из-за этого пациенты нередко выпускают предметы из рук, например разбивают кружки и роняют зубные щётки. Однако возможны различные вариации миоклоний.
Приступы учащаются в утренние часы, особенно при недосыпе или вынужденном пробуждении.
В 90 % случаев, помимо миоклонических эпизодов, отмечаются и генерализованные судорожные приступы [6] . После серии вздрагиваний в патологический процесс часто вовлекаются обе стороны тела.
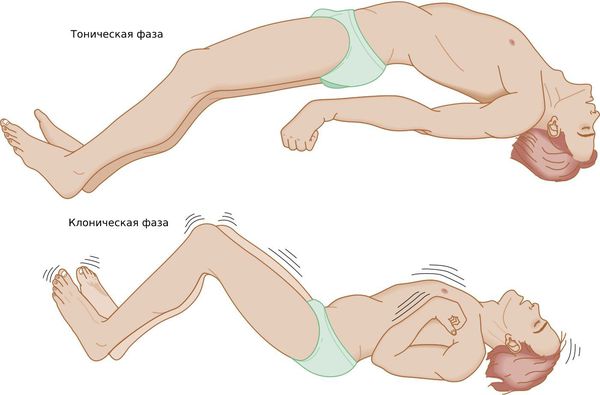
Генерализованный тонико-клонический приступ — состояние, при котором полностью отключается сознание. Приступ начинается с тонической фазы: напряжения в мышцах и специфического вскрикивания или хрипения. Руки полусогнуты и приподняты вверх или прижаты к телу. В этот момент из-за спазма дыхательной мускулатуры меняется цвет лица: оно синеет или сереет.
Далее развивается клоническая фаза, которая проявляется ритмичными подёргиваниями в конечностях. Она завершается полным мышечным расслаблением.
Третий вид эпилептических приступов при юношеской миоклонической эпилепсии — это абсансы [8] . Во время эпизода больной застывает, его взгляд устремлён в одну точку, сознание отключено. Состояние длится до 15 секунд и часто воспринимается окружающими как задумчивость. Сами пациенты могут не замечать эти приступы или воспринимать их как "провалы в памяти".
Патогенез юношеской миоклонической эпилепсии
Мозг человека состоит из двух основных типов клеток: нейронов и глии. Нейроны — это электрически возбудимые клетки, которые обрабатывают, хранят и передают информацию с помощью электрических и химических сигналов. Глиальные клетки играют в этом процессе вспомогательную роль.
Нейроны могут соединяться друг с другом и образовывать нервные сети. В пределах одного нейрона и его отростков информация передаётся в виде электрического возбуждения. В синапсе (месте контакта между нервными клетками) оно приводит к выделению различных химических веществ — нейромедиаторов.
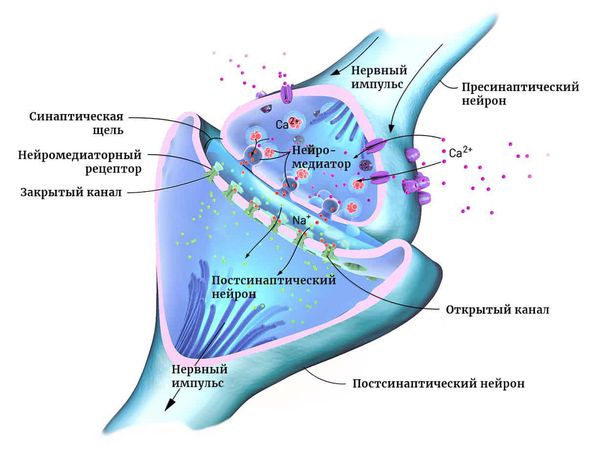
Нейромедиатор взаимодействует с рецепторами на мембране следующего нейрона. В результате в нём возникает электрическое возбуждение. Или не возникает — это зависит от конкретного нейромедиатора, активного в данный момент.
Чтобы заряд менялся в сторону возбуждения, в клетку должны поступать положительно и отрицательно заряженные ионы. Они проходят через ионные каналы в её мембране. Ионными каналами называют белки, образующие п о́ ру для обмена клетки с окружающей средой ионами K+, Na+ и другими [9] .
В нервных сетях между возбуждением и торможением работы нейронов поддерживается постоянный баланс. При сдвиге равновесия в сторону возбуждения происходит эпилептический приступ.
К юношеской миоклонической эпилепсии приводят мутации в генах ионных каналов. Однако выявлены нарушения и в других генах, также влияющих на процессы возбуждения в головном мозге [4] .
Классификация и стадии развития юношеской миоклонической эпилепсии
В Международной классификации болезней (МКБ-10) юношеская миоклоническая эпилепсия шифруется кодом G40.3 [10] .
В 2017 году Международная лига борьбы с эпилепсией (ILAE) обновила классификацию заболевания, выделив четыре уровня диагностики:
1. Определить тип приступа: фокальный (возникающий из одного очага), генерализованный и с неизвестным началом. Миоклонические, тонико-клонические приступы и абсансы относятся к генерализованным приступам.
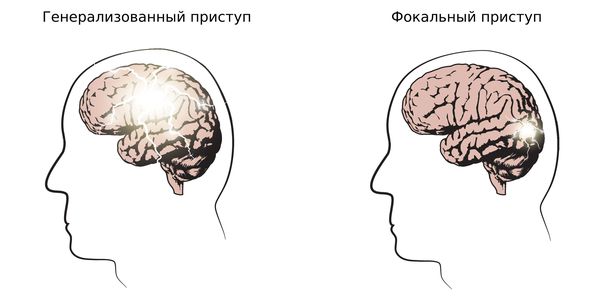
2. Установить тип эпилепсии: фокальная, генерализованная, сочетанная (фокальная + генерализованная) и неизвестная. Юношеская миоклоническая эпилепсия относится к генерализованной эпилепсии.
3. Определить эпилептический синдром. Юношеская миоклоническая эпилепсия как раз и является синдромом. Синдром включает типы приступов, возраст дебюта заболевания, характерные изменения на ЭЭГ, провоцирующие факторы и зачастую прогноз заболевания. Все эти факторы определяют лечебную тактику [11] .
4. Выявить причины заболевания: генетические, структурные, метаболические, иммунные, инфекционные и с неизвестной этиологией. Юношеская миоклоническая эпилепсия в большинстве случаев вызвана генетическими факторами.
Классификация юношеской миоклонической эпилепсии проводится в зависимости от течения заболевания. Главный критерий — это наличие миоклонических приступов. Также выделяют варианты течения с добавлением генерализованных судорожных приступов и/или абсансов.
Осложнения юношеской миоклонической эпилепсии
Пациенты часто не обращают внимания на патологические сокращения мышц, поэтому к неврологу и эпилептологу больной зачастую обращается после появления генерализованных тонико-клонических приступов. В результате противоэпилептические препараты назначают с опозданием. На фоне этого приступы могут учащаться и угрожать здоровью и жизни пациента травмами и утоплениями.
Серьёзным осложнением эпилепсии, в том числе и юношеской миоклонической эпилепсии, является внезапная смерть (SUDEP — Sudden Unexpected Death EPilepsy). Среди людей, страдающих эпилепсией, риск внезапной смерти в 20 раз выше, чем среди населения в целом [12] .
Точные причины SUDEP не установлены. Предполагается, что гибель пациентов связана с нарушением дыхания и развитием аритмии после приступа. Вероятность внезапной смерти при эпилепсии повышается при наличии генерализованных тонико-клонических приступов. Также важно, когда заболевание проявилось и сколько оно длится [12] .
При наличии дневных генерализованных приступов в течение предыдущего года риск развития SUDEP возрастает в 27 раз, ночных — в 15 раз. Проживание в одиночестве повышает риск внезапной смерти в 5 раз. Также SUDEP чаще встречается при злоупотреблении психоактивными веществами и алкоголем [13] .
Снизить риск внезапной смерти при эпилепсии можно, если придерживаться назначенного лечения: не пропускать приём противоэпилептических препаратов, не менять самостоятельно его частоту и дозировку [14] [15] .
Диагностика юношеской миоклонической эпилепсии
Основной диагностический критерий заболевания — это наличие миоклонических приступов.
Сбор анамнеза
На приёме врач спрашивает о необычных внезапных состояниях:
- вздрагиваниях в теле;
- дежавю — состоянии, при котором человек ощущает, что когда-то уже был в подобной ситуации или месте;
- потере сознания и т. д.
Пациенты могут не обращать внимания на такие симптомы и считать их своей особенностью. Абсансы и генерализованные тонико-клонические приступы с потерей сознания, особенно во сне, они могут и вовсе забывать. Поэтому при сборе анамнеза важно выяснить обстоятельства приступа не только у самих пациентов, но и у родственников и очевидцев.
Электроэнцефалограмма (ЭЭГ)
Основным способом диагностики эпилепсии является электроэнцефалограмма — метод исследования, при котором регистрируется суммарная электрическая активность клеток коры головного мозга.
Сейчас диагноз "эпилепсия" устанавливают с помощью длительного видео-ЭЭГ мониторинга — электроэнцефалограмма записывается параллельно с одной или несколькими видеокамерами, датчиком ЭКГ и при необходимости дополнительным контролем мышечной активности, частоты и глубины дыхания.
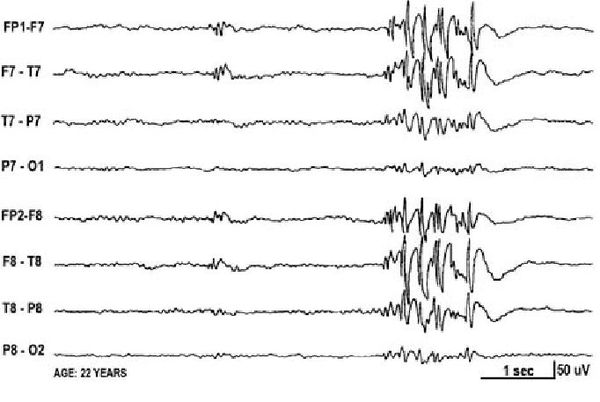
Основной фон биоэлектрической активности при юношеской миоклонической эпилепсии, как правило, соответствует возрастной норме. Патологическая активность проявляется короткими и генерализованными разрядами полиспайков (островолновых комплексов), которые регистрируются при миоклонических вздрагиваниях и полипик-волновыми комплексами между приступами.
При заболевании часто встречается феномен фотосенситивности. Для её выявления во время ЭЭГ пациента просят закрыть глаза и проводят ритмичную фотостимуляцию с частотой около 15 Гц [16] .
Эпилептическая фотосенситивность — это предрасположенность к приступам под влиянием света. Может протекать бессимптомно или проявляться эпилептическими приступами под воздействием провоцирующих факторов: видеоигр, работы за компьютером, просмотра телевизора, мигающего освещения в ночных клубах и света природного происхождения.
На МРТ патологические изменения в головном мозге при юношеской миоклонической эпилепсии не выявляются [17] .
Интеллект и неврологический статус при заболевании находятся в норме. Выражена эмоциональная неустойчивость и признаки невротического развития личности: резкая смена настроения, вспыльчивость и повышенная тревожность
Лечение юношеской миоклонической эпилепсии
Образ жизни
При эпилепсии следует соблюдать режим сна и бодрствования, исключить алкоголь и избегать резких вспышек света. Также нужно по возможности уменьшить стрессы, переживания и тревоги [20] .
Антиэпилептические препараты
Приём антиэпилептических препаратов (АЭП) позволяет устранить до 90 % приступов. Монотерапия (лечение одним препаратом) при юношеской миоклонической эпилепсии применяется в 79 % случаев, дуотерапия (двумя препаратами) — 17 %, политерапия (несколькими препаратами) — 4 % [16] .
Прекращать приём лекарств рекомендуется не ранее чем через пять лет полной клинико-нейрофизиологической ремиссии. Но даже спустя 4–7 лет ремиссии рецидивы после отмены терапии возникают у 70 % больных. Поэтому пациентам с юношеской миоклонической эпилепсией может быть рекомендован пожизненный приём АЭП [21] .

Ранее лидерами в лечении юношеской миоклонической эпилепсии являлись препараты вальпроевой кислоты. Они эффективны для прекращения приступов, но вызывают много побочных эффектов:
Также выявлено, что они обладают повышенным тератогенным эффектом по сравнению с другими АЭП. Тератогенное действие — это нарушение эмбрионального развития ребёнка при приёме препаратов матерью. Поэтому назначение вальпроатов, в особенности у молодых женщин, ограничено [18] .
В настоящее время препаратом выбора стартовой терапии является "Леветирацетам". Он хорошо переносится и эффективно устраняет все три вида приступов, в том числе сопровождающихся фотосенситивностью [19] .
Также используется препарат "Ламотриджин". Он эффективно подавляет генерализованные тонико-клонические судороги и абсансы, но в половине случаев способствует учащению миоклоний. Его применение в монотерапии у пациентов с частыми миоклоническими приступами ограничено, но лекарство можно использовать в комбинированной терапии [21] .
Помимо перечисленных препаратов, могут применяться "Топирамат", "Зонисамид", "Перампанел" и "Фенобарбитал".
Чтобы избежать учащения приступов и усиления симптомов, важно ограничить приём "Карбамазепина", "Окскарбазепина", "Фенитоина", "Габапентина" и "Вигабатрина". Эти лекарства могут повышать гипервозбудимость мембраны клеток головного мозга, что приводит к обострению состояния [21] .
Прогноз. Профилактика
Прогноз определяется индивидуально в зависимости от частоты приступов, эффективности АЭП, возраста начала заболевания и т. д. Лечение часто не помогает пациентами с тремя видами приступов [21] .
Без приёма противоэпилептических препаратов (АЭП) приступы могут сохраняться всю жизнь. Их частота, как правило, снижается только после 40 лет [20] .
Эффективность АЭП в предотвращении приступов достигает 90 %. При отмене терапии часто возникают рецидивы, поэтому потребуется длительный приём препаратов, иногда пожизненный.
Качество жизни значительно ухудшается при частых миоклонических и генерализованных тонико-клонических приступах, при которых пациенты рискуют получить травмы.
Профилактика
Особое внимание стоит уделить образу и режиму жизни пациента. Самыми мощными провоцирующими факторами являются недосыпание и злоупотребление алкоголем. А учитывая, что дебют заболевания приходится на подростковый возраст, молодые люди часто нарушают эти рекомендации, особенно в студенческие годы.
Пациент, у которого выявили фотосенситивность, предрасположен к приступам под воздействием мерцающего света. Поэтому им необходимо ограничить просмотр телевизора и работу за компьютером, исключить видеоигры и избегать посещения ночных клубов.
У всех пациентов с эпилепсией имеются определённые социальные ограничения: они не могут работать в некоторых сферах, водить автомобиль и нести военную службу. Все они определяются индивидуально соответствующими комиссиями.
Читайте также: