
Вич и гемолитическая анемия
Обновлено: 19.04.2024
Первичный иммунодефицит (ПИД) — это гетерогенная группа редких, преимущественно наследственных заболеваний, характеризующихся нарушением работы одного или нескольких компонентов иммунной системы.
Такие нарушения иммунитета приводят к повышенной уязвимости организма к различным патогенам, выражающейся в частых и рецидивирующих инфекционных заболеваниях, развитии хронических и системных болезней (аутоиммунных, онкологических).
Причины
В отличие от вторичных иммунодефицитов, возникающих в результате развития хронических заболеваний, иммуносупрессии, вызванной приемом лекарств, наличия ВИЧ-инфекции, естественного старения или недостаточности питания, большинство случаев первичного иммунодефицита ассоциированы с генетическими нарушениями. Дефектные гены передаются по наследству ребенку от одного или обоих родителей.
На данный момент известны более 350 первичных иммунодефицитов, почти все они ассоциированы с генетическими нарушениями. При этом до 70-90% людей, живущих с ПИД, не диагностированы, особенно в регионах с низкой доступностью лабораторного и генетического тестирования.
Первичные иммунодефициты классифицируют в зависимости от того, какая часть многокомпонентной иммунной системы поражена:
- дефицит гуморального иммунитета (B-клеток или B-лимфоцитов — типа лейкоцитов, вырабатывающих антитела, иммуноглобулины);
- дефицит клеточного иммунитета (T-клеток или T-лимфоцитов — типа лейкоцитов, помогающих идентифицировать и уничтожать чужеродные или аномальные клетки);
- комбинированный дефицит гуморального и клеточного иммунитета (B-лимфоцитов и T-лимфоцитов);
- дефекты фагоцитоза;
- дефицит белков комплемента (защитных белков, помогающих клеткам иммунной системы выявлять и уничтожать чужеродных агентов);
- аутовоспалительные заболевания (например, интерферонопатии 1 типа);
- иммунная дисрегуляция;
- фенокопии ПИД (не наследуемые, а приобретенные в процессе жизни) — ассоциированные с соматическими мутациями или появлением аутоантител.
Распространенность ПИД варьируется в зависимости от типа патологии. Более половины ПИД связаны с недостаточностью B-лимфоцитов. Примерно две трети пациентов — дети.
Симптомы
Клиническая картина первичных иммунодефицитов весьма разнородна и зависит от типа расстройства, она может включать не только иммунологические нарушения, но и желудочно-кишечные расстройства, гематологические и аутоиммунные заболевания, атопию, злокачественные новообразования. Кроме того, заболевания каждой группы ПИД имеют частично совпадающие симптомы и общие признаки типичных заболеваний.
Главной отличительной чертой ПИД являются частые, длительные, рецидивирующие инфекции, тяжело поддающиеся лечению, а также т. н. оппортунистические инфекции, которые для человека без иммунодефицита не опасны.
Симптомы первичного иммунодефицита могут включать:
- частые и рецидивирующие пневмонии, бронхиты, инфекции носовых пазух, дыхательных путей, уха, ротовой полости, кожные инфекции, менингит;
- задержку роста и развития;
- желудочно-кишечные расстройства (хроническая диарея, недостаточность питания, мальабсорбция, хронический лямблиоз, ВЗК, атрофический гастрит);
- аутоиммунные и аутовоспалительные заболевания (аутоиммунный тиреоидит, ревматоидный артрит, системная красная волчанка, дерматомиозит, диабет 1 типа);
- гематологические нарушения (аутоиммунная гемолитическая анемия, нейтропения, тромбоцитопения).
У некоторых пациентов с ПИД может наблюдаться целый комплекс связанных признаков заболевания. Например, для пациентов с хронической гранулематозной болезнью характерны рецидивирующие бактериальные или грибковые инфекции, а также хроническое воспаление ЖКТ и дыхательных путей. Для пациентов с синдромом Вискотта — Олдрича (Х-сцепленное рецессивное заболевание) характерны такие проявления, как экзема, рецидивирующие бактериальные инфекции и тромбоцитопения.
По сравнению с людьми со здоровой иммунной системой, у пациентов с ПИД выше риск злокачественных новообразований: лимфомы, лейкемии, опухолей пищеварительного тракта и вирус-индуцированного рака.
Диагностика
Первичные иммунодефициты достаточно сложно диагностировать и дифференцировать — в связи с неоднородным характером клинических проявлений и их схожестью с симптомами типичных болезней (синусита, бронхита, пневмонии, гастроэнтерита, менингита).
Иммунологические нарушения подтверждаются лабораторными исследованиями. Например, может проводиться оценка гуморального иммунитета и клеточного иммунитета, фагоцитарной функции, функции NK-клеток (естественных киллеров), системы комплемента. Необходимость в конкретных лабораторных исследованиях (оценке уровня сывороточных иммуноглобулинов, количественном измерении иммуноглобулинов, количественном измерении T-лимфоцитов, определении внутриклеточных цитокинов и белков, пролиферативной активности, клеточного цикла, цитотоксичности и др.) определяет иммунолог, в зависимости от клинической картины.
Генетическое тестирование (хромосомный анализ, флуоресцентная гибридизация in situ, полное секвенирование генома и экзома) играет важную роль для подтверждения диагноза, лечения и прогноза. Критерии выбора метода генетического тестирования основаны на стремлении к точной и быстрой постановке диагноза с минимальной финансовой нагрузкой для пациента.
Лечение первичного иммунодефицита
Лечение ПИД включает профилактику и контроль рецидивирующих инфекций, устранение основной причины иммунного нарушения, терапию заболеваний с ним связанных (аутоиммунных или онкологических).
Для профилактики и лечения частых и рецидивирующих инфекций назначается курс антибиотикотерапии (только инфекции, вызванные бактериями или грибками), прицельная (!) противовирусная терапия и симптоматическая терапия.
Для коррекции иммунитета могут назначаться иммуноглобулинотерапия (восполнение дефицита конкретных антител), интерферон-гамма терапия (синтетический аналог интерферонов, блокирующих репликацию вируса и стимулирующих клетки иммунной системы, используется для лечения хронической гранулематозной болезни, одной из форм ПИД), терапия факторами роста (при ПИД, вызванном недостатком лейкоцитов).
Защитный иммунитет пациентов с ПИД, более подверженных риску инфекции, обеспечивает регулярная и своевременная иммунизация, однако живые вакцины (от полиемиелита, кори, краснухи и паротита) для детей с первичными иммунодефицитами подходят не всегда.
Трансплантация костного мозга или трансплантация гемопоэтических стволовых клеток может рассматриваться как вариант лечения жизнеугрожающего ПИД, в частности тяжелого комбинированного иммунодефицита (или тяжелой комбинированной недостаточности — ТКИН). В последние годы достигнут значительный прогресс в лечении ТКИН (аденозиндезаминаза-ТКИН, X-сцепленный ТКИН, хроническая гранулематозная болезнь, синдром Вискотта — Олдрича) с помощью генной терапии. Переливание генно-скорректированных клеток показывает многообещающие результаты, однако пока метод применяют очень осторожно, поскольку такое лечение может приводить к неконтролируемому развитию у пациентов лейкемии.
Прогноз для пациентов с ПИД варьируется в зависимости от иммунных нарушений и тяжести течения заболевания. К сожалению, без трансплантации костного мозга или гемопоэтических стволовых клеток дети с тяжелой комбинированной недостаточностью умирают в течение первых 2 лет жизни. У детей, перенесших трансплантацию в возрасте до 3 месяцев, прогноз улучшается. У пациентов с менее тяжелыми формами ПИД, получивших адекватное лечение, показатели долгосрочной выживаемости и качества жизни намного выше.
Особенности и преимущества лечения первичного иммунодефицита в клинике Рассвет
Если у вашего ребенка или у вас наблюдаются частые или рецидивирующие инфекции с тяжелым течением, обязательно запишитесь на прием к педиатру или терапевту Рассвета. При подозрении на первичный (или вторичный) иммунодефицит эти специалисты направят вас к иммунологу.
Научно обоснованные и доказанно эффективные методы диагностики и лечения первичного иммунодефицита, которые используют иммунологи Рассвета, позволят вам сохранить хорошее качество жизни и здоровье надолго.
Наши врачи-иммунологи помогут исключить или выявить и устранить иммунодефицит, при необходимости вашим лечением займется мультидисциплинарная команда специалистов — терапевты, пульмонологи, ревматологи.
ЧТО ВЫЗЫВАЕТ АНЕМИЮ?
Костный мозг производит красные кровяные тельца. Этот процесс требует наличия железа, витамина В12 и фолиевой кислоты (витамина В6). Эритропоэтин стимулирует производство красных кровяных телец. Эритропоэтин это гормон, вырабатываемый почками.
Анемия может быть вызвана тем, что организм вырабатывает недостаточное количество красных кровяных телец. Также это может быть вызвано их разрушением, гибелью. Несколько факторов могут вызывать анемию:
• Нехватка железа, витамина В12 или фолиевой кислоты. Нехватка фолиевой кислоты может вызвать мегалобластическую анемию, при которой красные кровяные тельца становятся большими но бледными (см. информационный листок 121).
• Повреждения костного мозга или почек
• Потеря крови в связи с внутренним кровотечением или менструальным циклом у женщин.
• Разрушение красных кровяных телец (гемолитическая анемия)
ВИЧ - инфекция может вызывать анемию. Также это могут делать и оппортунистические инфекции (см. информационный листок 500) т.н. ВИЧ-ассоциированные заболевания. Многие препараты широко применяющиеся для лечения ВИЧ и связанных с ним заболеваний могут вызывать анемию.
АНЕМИЯ И ВИЧ
Раньше анемия была гораздо более распространена. У более чем 80% людей с диагнозом СПИД была та или иная степень анемии. У людей, с более развитой ВИЧ – инфекцией или низким уровнем CD4 клеток, наблюдались более высокие уровни анемии.
Уровень анемии начал снижаться, когда люди стали применять комбинированную антиретровирусную терапию (АРВТ). Тяжелые формы анемии стали редки. Однако, АРВТ не уничтожила анемию. Было проведено большое исследование, которое показало что около 46% пациентов имели мягкую или среднюю формы анемии, после года приема АРВТ.
Несколько факторов связаны с повышенной степенью анемии среди людей с ВИЧ:
• Низкий уровень CD4 клеток (см. информационный листок 124)
• Высокая вирусная нагрузка (информационный листок 125)
• Прием АЗТ (Ретровир, информационный листок 411)
• Быть африканцем
• Быть женщиной
ВИЧ быстрее прогрессирует в людях с диагнозом анемия. Этот показатель примерно в пять раз больше среди людей с анемией, по сравнению с теми, у кого ее нет. Анемия также связана с высоким риском смерти. Лечение анемии, по имеющимся сведениям, исключает такие риски.
КАК ЛЕЧИТСЯ АНЕМИЯ?
Способ лечения анемии зависит от причины ее возникновения.
• Во-первых, лечат все хронические кровотечения. Это может быть внутреннее кровотечение, геморрой или даже частое носовое кровотечение.
• Далее, корректируется любая нехватка железа, витамина В12 или В6 в организме.
• Прекращается прием, или снижается доза препарата, вызывающего анемию
Эти методы могут не сработать. Например, невозможно будет прекратить принимать все лекарства, вызывающие анемию. Есть два дополнительных метода лечения –это инъекции эритропоэтина и переливание крови.
Эритропоэтин стимулирует производство красных кровяных телец. В 1985 году, ученые нашли способ производить синтетический эритропоэтин. Он вводится подкожно, обычно раз в неделю. Наиболее известные брэнд эритропоэтина –это Прокрит (Procrit®).
Большое исследование среди ЛЖВ выявило, что инъекции эритропоэтина понижают риск смерти. Переливание крови же, как кажется, увеличивает его. В связи с такими рисками, переливания крови редко используют для лечения анемии.
Переливание крови ранее было единственным способом лечения тяжелых форм анемии. Однако, переливание может вызвать инфекцию и подавить иммунную систему. Переливание, как кажется, вызывает быстрое прогрессирование ВИЧ и увеличивает риск смерти у пациентов с ВИЧ.
Анемия повышает утомляемость и при ней люди чувствуют себя плохо. Она увеличивает риск прогрессирования заболевания и смерти. Она может быть вызвана ВИЧ-инфекцией или другими заболеваниями. Многие препараты использующиеся для лечения ВИЧ и ассоциированных с ним заболеваний также могут вызывать анемию.
Анемия всегда была проблемой для людей с ВИЧ/СПИД. Количество серьезных, тяжелых случаев анемии невероятно снизилось, когда люди начали использовать АРВТ. Однако, почти у половины ЛЖВ все ещё наблюдается мягкая или средняя форма анемии.
Лечение анемии улучшает здоровье и выживаемость ЛЖВ. Лечение кровотечений, нехватки железа, или витаминов –это первые шаги. Если возможно, необходимо прекратить прием препаратов, вызывающих анемию. Если необходимо, пациент должен получать лечение эритропоэтином, или в крайне редких случаях, переливание крови.
Аутоиммунная гемолитическая анемия (АИГА) — группа редких приобретенных гематологических болезней и синдромов, характеризующихся гемолизом (разрушением) эритроцитов, вследствие образования аутоантител к антигенам этих компонентов крови.
АИГА может возникнуть в любом возрасте, чаще ей подвержены женщины (60%). Патология развивается постепенно или внезапно.
Выделяют два основных типа АИГА: тепловую (аутоантитела наиболее активны/атакуют эритроциты при температуре 37-40 °C) и холодовую — с холодовыми гемолизинами (аутоантитела наиболее активны при температуре менее 30 °C, эритроциты разрушаются даже при локальном воздействии холода, например, когда человек пьет холодную воду или моет руки в холодной воде).
Причины
Причины возникновения заболевания изучены недостаточно. На сегодняшний день известно, что примерно 50% случаев т-АИГА являются идиопатическими (развиваются спонтанно). Тогда как х-АИГА ассоциированы с другими заболеваниями или возникают с ними одновременно: аутоиммунными, онкологическими, инфекционными (системная красная волчанка, лимфолейкоз, неходжкинская лимфома, вирус Эпштейна — Барр, цитомегаловирус, микоплазменная пневмония, гепатит, ВИЧ). Играть роль в развитии АИГА может и прием определенных лекарств, например, препаратов пенициллинового ряда (лекарственно индуцированная АИГА).
Симптомы
У некоторых людей заболевание может протекать бессимптомно, особенно если АИГА развивается постепенно (разрушение эритроцитов не настолько масштабно). В основном же симптомы АИГА схожи с клиническими проявлениями других типов анемии (слабость, повышенная утомляемость, бледность кожных покровов).
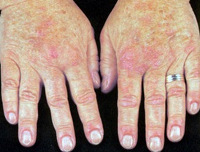
Симптомы при более тяжелом течении заболевания (быстром разрушении эритроцитов) включают желтушность кожных покровов и видимых слизистых оболочек, слабость, повышенную утомляемость с тахикардией и одышкой при физической нагрузке, ощущение дискомфорта в животе, чувство переполнения, холодные конечности и периферический цианоз (при холодовой АИГА). При физикальном осмотре выявляется спленомегалия (патологическое увеличение размеров селезенки) различной степени.
Если АИГА развивается на фоне другой патологии, преобладать могут симптомы основного заболевания, например: увеличение и болезненность лимфатических узлов, лихорадка, сильные боли в спине и ногах, головные боли, рвота, диарея, изменение цвета мочи на темно-коричневый.
Диагностика
Диагностика АИГА включает физикальный осмотр, общий и биохимический анализы крови, морфологическое исследование эритроцитов. Основным критерием диагностического поиска является определение аутоантител с помощью прямого антиглобулинового теста (прямой пробы Кумбса).
Лечение аутоиммунной гемолитической анемии
Первой линией терапии тепловой аутоиммунной гемолитической анемии являются глюкокортикостероиды. Высокодозная монотерапия преднизолоном может назначаться в течение 3-6 недель с постепенным снижением дозы или отменой в дальнейшем. Однако из-за сильно выраженных побочных эффектов такого лечения все чаще используют краткосрочную пульс-терапию метилпреднизолоном или дексаметазоном. Примерно треть пациентов достигают ремиссии, остальным же необходима поддерживающая терапия ГКС.
При неэффективности стероидной терапии рассматривается возможность проведения спленэктомии (хирургическое удаление селезенки), биологической терапии (ритуксимаб) или иммуносупрессивной терапии (циклоспорин, азатиоприн и др.).
Прогноз зависит от первопричины заболевания, своевременно начатого и правильно подобранного лечения. Благоприятный прогноз (достижение ремиссии, длительная ремиссия) полностью связан с положительным терапевтическим ответом у пациента и отсутствием осложнений.
Пациентам с холодовой аутоиммунной гемолитической анемией рекомендуется избегать триггеров (холода, в том числе инфузий холодных растворов), а также пройти лечение основного заболевания (например, лимфомы), если с ним связано развитие х-АИГА. Терапия первой линии при х-АИГА — ритуксимаб. ГКС не является терапией выбора из-за низкого терапевтического ответа у пациентов. Также неэффективна и спленэктомия. В тяжелых случаях рекомендован плазмоферез.
Особенности и преимущества лечения аутоиммунной гемолитической анемии в клинике Рассвет
Аутоиммунная гемолитическая анемия достаточно редкое заболевание крови, по мере его изучения критерии диагностики и лечения дорабатываются и обновляются.
Гематологи клиники Рассвет придерживаются стандартизированных диагностических критериев и терапевтических подходов, разработанных Международной консенсусной группой по АИГА. Мы осознаем важность точного диагностического поиска у таких пациентов, поскольку течение заболевания и эффективное лечение зависят от типа задействованных антител. Для диагностики первичной АИГА мы используем моноспецифический прямой антиглобулиновый тест, обязательно выясняем причины манифестации вторичной АИГА, т. е. выявляем основное заболевание, с которым связано развитие гемолитической анемии.
При лечении т-АИГА мы используем глюкокортикостероиды. Ритуксимаб назначаем только на раннем этапе тяжелого течения заболевания, а также при отсутствии быстрого терапевтического ответа на стероиды. Ритуксимаб в сочетании с антинеопластическим препаратом назначаем пациентам с х-АИГА в случаях, если их состояние требует лечения по клиническим признакам.
Гематология Рассвета представлена врачами высокой квалификации, имеющими большой опыт в выявлении и лечении сложнодиагностируемых и редких болезней. В своей работе наши специалисты используют международные протоколы лечения, применяют только безопасные, доказанные и эффективные методы.
Иммунные гемолитические анемии – группа заболеваний, обусловленных повышенным разрушением эритроцитов вследствие выработки антител против неизмененных красных кровяных телец или гаптенов, появившихся на мембране эритроцита. Различают изоиммунные, трансиммунные, гетерогенные и аутоиммунные гемолитические анемии. Клинические признаки: бледность или желтушность кожных покровов, умеренное увеличение печени и селезенки, боли в поясничной области, одышка и другие симптомы. Диагностика основана на изучении клинических данных, результатов лабораторных и инструментальных исследований. Лечение: гемотрансфузии, введение препаратов крови и кортикостероидов, иногда – спленэктомия.
Общие сведения
Иммунные гемолитические анемии – группа заболеваний, характеризующихся повреждением и преждевременной гибелью эритроцитов или эритрокариоцитов в связи с развитием иммунной реакции с участием IgG и IgM или иммунных лимфоцитов. Основные факторы, вызывающие разрушение эритроцитов – аутоиммунный процесс, гемотрансфузионные осложнения, эритробластоз плода и гемолиз, обусловленный действием некоторых лекарственных средств.
Гемолиз может происходить в самом кровеносном русле или вне сосудов: в печени, селезенке, костном мозге. В результате массовой гибели красных кровяных телец развиваются анемический и желтушный синдромы, свидетельствующие о нарушении функции печени, почек, дыхательной системы, системы кровообращения, других органов и систем организма. По данным статистики, распространенность иммунных гемолитических анемий составляет примерно 1 случай на 70-80 тысяч населения.
Причины иммунных гемолитических анемий
Возникновение этой группы заболеваний связано с воздействием различных неблагоприятных факторов внешней и внутренней среды, приводящих к развитию иммунных реакций против собственных эритроцитов. Чаще всего встречается аутоиммунный механизм, при котором происходит выработка антител против неизмененных естественных антигенов мембраны эритроцитов, находящихся в кровяном русле, или их предшественников – эритрокариоцитов костного мозга. Первичный причинный фактор, вызывающий разрушение эритроцитов, неизвестен (идиопатическая форма).
При вторичных анемиях патологический процесс развивается на фоне хронического лимфолейкоза, лимфомы, антифосфолипидного синдрома или иммунодефицитных состояний. Чаще встречается тепловая форма аутоиммунной анемии, при которой внутренняя среда организма имеет нормальные температурные параметры, а на эритроцитах располагаются иммуноглобулины класса G и компоненты комплемента C3 и C4. Красные кровяные тельца разрушаются макрофагами в печени и селезенке. Менее распространенная холодовая форма может быть идиопатической или вторичной, связанной с инфекцией (инфекционным мононуклеозом и микоплазменной пневмонией), переохлаждением и лимфопролиферативными заболеваниями у больных старше 60 лет. Реакция антиген-антитело с гемолизом развивается в периферическом кровяном русле, где температура опускается ниже 32 градусов.
Иммунная гемолитическая анемия может возникать при фиксации на мембране эритроцита фрагмента, имеющего лекарственное, вирусное или бактериальное происхождение (гетероиммунная форма). Образовавшиеся гаптены превращают красные кровяные тельца в чужеродные клетки-мишени для иммунной системы, что в итоге приводит к гемолизу. Чаще всего такую реакцию вызывают антибиотики из группы пенициллинов, цефалоспоринов и тетрациклинов, противотуберкулезные препараты, анальгетики и антиаритмические средства.
Изоиммунная форма встречается при несовместимости крови матери и плода по группе крови или резус-фактору. При этом антиэритроцитарные антитела матери через плаценту проникают к плоду и вызывают разрушение эритроцитов. Подобный механизм наблюдается и при переливании (гемотрансфузии) несовместимой крови от донора пациенту.
Классификация иммунных гемолитических анемий
Выделяют изоиммунные, трансиммунные, гетерогенные и аутоиммунные гемолитические анемии.
- К изоиммунным вариантам заболевания относятся посттрансфузионные осложнения, связанные с непереносимостью крови донора или неправильным соблюдением процедуры трансфузии, а также гемолитическая болезнь новорожденных.
- При трансиммунных анемиях гемолиз возникает при попадании к плоду через плаценту антител от матери, болеющей аутоиммунной анемией.
- Гетероиммунная (гаптеновая) анемия – результат изменения мембраны эритроцита при воздействии вирусов или лекарственных препаратов.
- Аутоиммунные гемолитические анемии (с тепловыми и холодовыми агглютининами).
Симптомы иммунных гемолитических анемий
Болезнь чаще начинается остро: повышается температура тела, появляется озноб, головная боль и головокружение, одышка, боли в эпигастральной области и пояснице. Кожные покровы становятся бледными, затем желтушными, могут появиться геморрагии, увеличивается селезенка, печень, цвет мочи становится темным.
При холодовой форме иммунной гемолитической анемии нередко нарушается периферическое кровообращение с развитием синдрома Рейно, посинением и отечностью кожных покровов кистей рук, лица, ушных раковин. В некоторых случаях нарушение кровоснабжения тканей конечностей может привести к развитию гангрены пальцев стоп.
Тяжелое течение эритробластоза плода характеризуется развитием ядерной желтухи с поражением центральной нервной системы – билирубиновой энцефалопатии. При этом отмечается вялость, заторможенность, снижение аппетита, частые срыгивания, судорожный синдром. При пальпации обнаруживают увеличение селезенки, печени, периферических лимфоузлов.
Диагностика иммунных гемолитических анемий
Чтобы установить правильный диагноз, определить форму заболевания, необходимо тщательное обследование пациента со сбором анамнеза, проведением клинического физикального осмотра врачом-гематологом, аллергологом-иммунологом, инфекционистом, ревматологом и другими специалистами. Уже на этапе осмотра можно обнаружить бледность, желтушность кожных покровов и слизистых, пропальпировать увеличенную селезенку и печень. Выраженность спленомегалии и гепатомегалии уточняется при проведении ультразвукового исследования (УЗИ печени и селезенки).
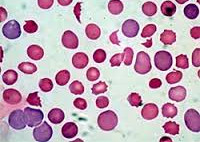
Лабораторная диагностика подтверждает наличие нормо- или гипохромной (реже – гиперхромной) анемии, ретикулоцитоза, увеличение СОЭ, гипербилирубинемию. В анализе мочи может выявляться протеинурия, уробилинемия, гемоглобинурия. При исследовании пунктата костного мозга обнаруживаются признаки гиперплазии за счет активации эритропоэза. При гемолитической болезни новорожденных выявляется выраженный эритробластоз (100-150 тысяч в 1 мкл).
Диагноз аутоиммунной гемолитической анемии подтверждается положительной прямой пробой Кумбса (прямым антиглобулиновым тестом) или полибреновым тестом (сенсибилизированная проба Кумбса). Дифференциальная диагностика проводится с другими иммунными болезнями, различными формами анемий, гемобластозами, тяжелыми отравлениями, болезнями печени и почек.
Лечение иммунных гемолитических анемий
Лечебная тактика различается при различных формах болезни. При аутоиммунном характере гемолиза с тепловыми антигенами проводится введение высоких доз иммуноглобулина, кортикостероидов, иногда - циклофосфамида и других иммуносупрессивных препаратов. Возможно применение плазмафереза. При неэффективности консервативной терапии рекомендуется проведение спленэктомии. При холодовой иммунной гемолитической анемии используется введение моноклональных антител (ритуксимаба), плазмаферез, трансфузия индивидуально подобранных, отмытых и подогретых эритроцитов.
При эритробластозе плода проводится дезинтоксикационная терапия, переливание крови или эритроцитарной массы. При пострансфузионных осложнениях необходимо проведение неотложных противошоковых мероприятий, борьба с синдромом диссеминированного внутрисосудистого свертывания.
Гемолитическая анемия – патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для данной группы заболеваний типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.
МКБ-10


Общие сведения
Гемолитическая анемия (ГА) - малокровие, обусловленное нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Данная группа анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных когортах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. Патология характеризуется укорочением жизненного цикла эритроцитов и их распадом (гемолизом) раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).
Причины
Этиопатогенетическую основу наследственных гемолитических синдромов составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфофункциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды, среди которых:
- Аутоиммунные процессы. Образование антител, агглютинирующих эритроциты, возможно при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии). Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода.
- Токсическое действие на эритроциты. В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Вызывать разрушение клеток крови может прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков).
- Механическое повреждение эритроцитов. Гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Патогенез
Классификация
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные. Наследственные ГА включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз – болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) – анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) – анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии- анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные ГА подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия – б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные) – обусловлены воздействием антител
- токсические – анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- механические - анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Симптомы
Наследственные мембранопатии, ферментопении и гемоглобинопатии
Наиболее распространенной формой данной группы анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке. Манифестация микросфероцитарной ГА возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом - обтурационная желтуха. При микросфероцитозе во всех случаях увеличена селезенка, а у половины пациентов – еще и печень. Кроме наследственной микросфероцитарной анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Приобретенные гемолитические анемии
Среди различных приобретенных вариантов чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер. Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка. При некоторых формах аутоиммунных анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Осложнения
Каждый вид ГА имеет свои специфические осложнения: например, ЖКБ – при микросфероцитозе, печеночная недостаточность – при токсических формах и т.д. К числу общих осложнений относятся гемолитические кризы, которые могут провоцироваться инфекциями, стрессами, родами у женщин. При остром массивном гемолизе возможно развитие гемолитической комы, характеризующейся коллапсом, спутанным сознанием, олигурией, усилением желтухи. Угрозу жизни больного несут ДВС-синдром, инфаркт селезенки или спонтанный разрыв органа. Неотложной медицинской помощи требуют острая сердечно-сосудистая и почечная недостаточность.
Диагностика
Определение формы ГА на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки. Лабораторный диагностический комплекс включает:
- Исследование крови. Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. При аутоиммунных анемиях большое диагностическое значение имеет положительная проба Кумбса.
- Анализы мочи и кала. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина.
- Миелограмму. Для цитологического подтверждения выполняется стернальная пункция. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, порфирии, гемобластозы. Пациента консультируют гастроэнтеролог, клинический фармаколог, инфекционист и другие специалисты.
Лечение
Различные формы ГА имеют свои особенности и подходы к лечению. При всех вариантах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости – гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной анемии выполняется спленэктомия. Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям – введение антидотов.
Прогноз и профилактика
Течение и исход зависят от вида анемии, тяжести протекания кризов, полноты патогенетической терапии. При многих приобретенных вариантах устранение причин и полноценное лечение приводит к полному выздоровлению. Излечения врожденных анемий добиться нельзя, однако возможно достижение длительной ремиссии. При развитии почечной недостаточности и других фатальных осложнений прогноз неблагоприятен. Предупредить развитие ГА позволяет профилактика острых инфекционных заболеваний, интоксикаций, отравлений. Запрещается бесконтрольное самостоятельное использование лекарственных препаратов. Необходимо тщательная подготовка пациентов к гемотрансфузиям, вакцинации с проведением всего комплекса необходимых обследований.
4. Клинические рекомендации по диагностике и лечению аутоиммунных гемолитический анемий/ Цветаева Н.В., Никулина О.Ф. - 2014.
Читайте также: