
3д модель вируса гриппа
Обновлено: 05.05.2024
Вирус гриппа — это широко распространенный, легко передающийся и быстро эволюционирующий возбудитель заболевания. Симптомы гриппа могут напоминать признаки обычной простуды, однако болезнь чревата осложнениями, которые особенно опасны для маленьких детей, пожилых людей и тех, кто страдает хроническими заболеваниями. Новые штаммы гриппа возникают ежегодно. Штаммы — это разновидности вируса, отличающиеся друг от друга двумя поверхностными белками, которые быстро накапливают изменения и тем самым увеличивают их разнообразие. В случае одновременного заражения организма разными штаммами белки могут попадать в формирующиеся в этом процессе новые частицы в произвольных сочетаниях, что вновь ведет к росту разнообразия вирусов. Это, в свою очередь, вызывает дополнительные проблемы с лечением инфекции и увеличивает риск межвидового заражения.
Грипп встречается не только у людей, но и среди животных. Циркулируя в их популяциях, вирус нередко приобретает наиболее неприятные для человека свойства.
История эпидемий гриппа
Успехи в разработке противовирусных препаратов и профилактических мер, а также активный мониторинг распространения и возникновения новых штаммов гриппа позволяют сокращать число жертв и интенсивность эпидемий. Однако и сейчас в результате вызываемых им осложнений погибает от 250 до 500 тысяч человек в год.
Международный проект компании Visual Science"Зоопарк вирусов" — первая успешная попытка создать модели наиболее распространенных и опасных вирусов человека с разрешением до атомов.
Задача построения научно достоверной 3D-модели вируса не так тривиальна, как кажется на первый взгляд. Ни один из имеющихся на сегодняшний день научных подходов, будучи применён по отдельности, не позволяет получить изображение целой вирусной частицы в атомном или даже молекулярном разрешении. На молекулярном уровне вирусы представляют собой структуры огромного размера: один вирус — это комплекс сотен макромолекул (белков,нуклеиновых кислот, в ряде случаев липидов). Несмотря на это, вирусы слишком малы для того, чтобы их можно было досконально изучить, используя электронную и тем более оптическую микроскопию: эти инструментальные методы хорошо подходят для исследования строения существенно более крупных биологических объектов, например клеток. В частности, электронная микроскопия даёт возможность получить грубые изображения, на которых видны лишь контурывириона (этот метод не всегда позволяет увидеть даже крупные поверхностные белки).
Для изучения отдельных белков широко используются методы рентгеноструктурной кристаллографии и ядерного магнитного резонанса. Полученные с их помощью данные представляют собой пространственные координаты атомов, входящих в состав молекулы, в наиболее энергетически выгодном положении из возможных. В контексте задачи визуализации структуры вириона недостатки этого метода связаны с невозможностью исследования с его помощью более массивных объектов (он неприменим даже к небольшим комплексам нескольких белков) и трудностью изучения подвижных и связанных с мембранами компонентов вируса.
Таким образом, задача создания научно достоверных 3D-моделей напоминает сборку паззла, где в исходном наборе не хватает значительной части фрагментов, отдельные фрагменты являются неполными, а иллюстрация результата, к которому стремятся собирающие паззл исследователи, груба, и потому даёт только общее представление о конечной картине. Тем не менее, сотни работ разных авторов со всего мира проливают свет на многие вопросы структуры и морфологии компонентов вирионов, а также их взаимодействия. При тщательном анализе всех научных работ, с учетом мнений признанных экспертов из мировых научных центров и помощи специалистов Отдела молекулярного моделирования компании Visual Science, устраняющих пробелы в имеющихся исследованиях по молекулярной биологии, вирусологии и кристаллографии, появляется возможность создать максимально точные и достоверные модели вирусов, которые и представлены в проекте Viral Park.
Компоненты вируса: более 200 тысяч молекул 11 типов
Вирион гриппа имеет форму удлинённой по одной из осей сферы диаметром 80-120 нанометров, что в тысячи раз меньше толщины человеческого волоса. Продолговатую форму вируса определяет слой структурного белка — матрикса. С внешней стороны матрикс окружён мембраной с поверхностными белками (гемагглютинином и нейраминидазой) и ионными каналами. Геном вируса — это восемь молекул РНК в составе спиральных комплексов. Он, как и белки, необходимые для полноценной работы вируса в зараженной клетке, находится внутри частицы.
— Гемагглютинин связывается с рецепторами на поверхности клетки, позволяя частице слиться с мембраной клеточной везикулы, проникнуть внутрь нее и доставить геном вируса в цитоплазму.
— Нейраминидаза нужна для того, чтобы только что сформированные вирионы могли отделяться от клеточной мембраны и заражать другие клетки.
— Белок матрикса — одна из основных структурных молекул вируса. Именно с этим белком связана характерная форма вирусной частицы и расположение в ней внутренних компонентов (что, в свою очередь, детерминирует правильность распаковки и процесса заражения), он же определяет её размеры. Молекулы белка матрикса связываются как с поверхностными белками, так и с нуклеопротеиновыми комплексами вируса, позволяя всем компонентам попадать в частицу.
— Белок M2 образует каналы, через которые внутрь частицы проникают ионы водорода после того, как та оказывается в клетке. Это запускает разборку вируса и высвобождение его генома.
— Нуклеопротеин упаковывает фрагменты вирусного генома в компактные спирально закрученные комплексы геномной РНК и белка NP — рибонуклеопротеиды (РНП),которые помещаются внутри вирусной частицы.
— Белок ядерного экспорта обеспечивает транспорт копий РНК генома из ядра, где они образовались, к месту сборки новых частиц вируса у поверхности зараженной клетки.
— Полимеразный комплекс необходим для создания копий РНК вируса, одна часть которых нужна для того, чтобы синтезировать структурные белки, а другая — чтобы упаковываться в новые вирусные частицы. Геном вируса несет информацию, необходимую для синтеза вирусных белков. Он представлен восемью молекулами РНК, отличающимися длиной и набором кодируемых белков.
— Мембрана вируса формируется из мембраны клетки, в которой он образовался. В ее состав входят молекулы фосфатидилхолина, фосфатидилэтаноламина, сфингомиелина и холестерина — в характерных для клеток человека пропорциях.
Процесс создания научно достоверных 3Д моделей вирусов
Пространственные структуры некоторого количества белков, входящих в состав вирионов (этадоля варьируется от 25 до 50%), описаны не полностью: проблемы возникают из-за того, что не все белковые молекулы или отдельные их фрагменты возможно кристаллизовать (а это является условием проведения рентгеноструктурного анализа). Обычно в структурах недостаёт подвижных фрагментов молекул, а также трансмембранных участков и гликозильных остатков поверхностных белков. В то же время для комплексов и ансамблей белков в большинстве случаев неизвестно, какие поверхности компонентов формируют контакты друг с другом, а какие — нет. Предсказание и описание таких взаимодействий необходимо при построении любой вируснойчастицы (для большей части входящих в неё молекулярных комплексов и структур).
Эти проблемы позволяет решить структурная биоинформатика, основанная на вычислительных методах: в данном подходе используется предсказание пространственной структуры исследуемой молекулы на основе структур схожих или родственных протеинов и исходной последовательности аминокислот, которая в большинстве случаев известна заранее. Кроме того, подобные методы позволяют рассчитать меж- и внутримолекулярные взаимодействия. В компании Visual Science эту задачу решает Отдел молекулярного моделирования и динамики: работающие в нем специалисты — молекулярные биологи и биоинформатики, обладающие научными степенями. Они проводят анализ опубликованных ранее кристаллографических данных (параллельно оценивая точность описания похожих белков), осуществляют моделирование недостающих фрагментов и в результате проводят сборку полных достоверных моделей всех компонентов и комплексов вирусной частицы, комбинируя известные данные с теми, что получены ими в Отделе. Подобный уровень сотрудничества с научным сообществом, а также столь высокая степень вовлечения в этот процесс структурных биологов из числа сотрудников Отдела молекулярного моделирования компании на данный момент недоступны ни одной студии научной и медицинской визуализации в мире — кроме Visual Science.
Дополнительные материалы: научный обзор и панорама 3Д модели вируса гриппа.
Вирус гриппа — это широко распространенный, легко передающийся и быстро эволюционирующий возбудитель заболевания. Симптомы гриппа могут напоминать признаки обычной простуды, однако болезнь чревата осложнениями, которые особенно опасны для маленьких детей, пожилых людей и тех, кто страдает хроническими заболеваниями. Новые штаммы гриппа возникают ежегодно. Штаммы — это разновидности вируса, отличающиеся друг от друга двумя поверхностными белками, которые быстро накапливают изменения и тем самым увеличивают их разнообразие. В случае одновременного заражения организма разными штаммами белки могут попадать в формирующиеся в этом процессе новые частицы в произвольных сочетаниях, что вновь ведет к росту разнообразия вирусов. Это, в свою очередь, вызывает дополнительные проблемы с лечением инфекции и увеличивает риск межвидового заражения.
Грипп встречается не только у людей, но и среди животных. Циркулируя в их популяциях, вирус нередко приобретает наиболее неприятные для человека свойства.
История эпидемий гриппа
Успехи в разработке противовирусных препаратов и профилактических мер, а также активный мониторинг распространения и возникновения новых штаммов гриппа позволяют сокращать число жертв и интенсивность эпидемий. Однако и сейчас в результате вызываемых им осложнений погибает от 250 до 500 тысяч человек в год.
Международный проект компании Visual Science"Зоопарк вирусов" — первая успешная попытка создать модели наиболее распространенных и опасных вирусов человека с разрешением до атомов.
Задача построения научно достоверной 3D-модели вируса не так тривиальна, как кажется на первый взгляд. Ни один из имеющихся на сегодняшний день научных подходов, будучи применён по отдельности, не позволяет получить изображение целой вирусной частицы в атомном или даже молекулярном разрешении. На молекулярном уровне вирусы представляют собой структуры огромного размера: один вирус — это комплекс сотен макромолекул (белков,нуклеиновых кислот, в ряде случаев липидов). Несмотря на это, вирусы слишком малы для того, чтобы их можно было досконально изучить, используя электронную и тем более оптическую микроскопию: эти инструментальные методы хорошо подходят для исследования строения существенно более крупных биологических объектов, например клеток. В частности, электронная микроскопия даёт возможность получить грубые изображения, на которых видны лишь контурывириона (этот метод не всегда позволяет увидеть даже крупные поверхностные белки).
Для изучения отдельных белков широко используются методы рентгеноструктурной кристаллографии и ядерного магнитного резонанса. Полученные с их помощью данные представляют собой пространственные координаты атомов, входящих в состав молекулы, в наиболее энергетически выгодном положении из возможных. В контексте задачи визуализации структуры вириона недостатки этого метода связаны с невозможностью исследования с его помощью более массивных объектов (он неприменим даже к небольшим комплексам нескольких белков) и трудностью изучения подвижных и связанных с мембранами компонентов вируса.
Таким образом, задача создания научно достоверных 3D-моделей напоминает сборку паззла, где в исходном наборе не хватает значительной части фрагментов, отдельные фрагменты являются неполными, а иллюстрация результата, к которому стремятся собирающие паззл исследователи, груба, и потому даёт только общее представление о конечной картине. Тем не менее, сотни работ разных авторов со всего мира проливают свет на многие вопросы структуры и морфологии компонентов вирионов, а также их взаимодействия. При тщательном анализе всех научных работ, с учетом мнений признанных экспертов из мировых научных центров и помощи специалистов Отдела молекулярного моделирования компании Visual Science, устраняющих пробелы в имеющихся исследованиях по молекулярной биологии, вирусологии и кристаллографии, появляется возможность создать максимально точные и достоверные модели вирусов, которые и представлены в проекте Viral Park.
Компоненты вируса: более 200 тысяч молекул 11 типов
Вирион гриппа имеет форму удлинённой по одной из осей сферы диаметром 80-120 нанометров, что в тысячи раз меньше толщины человеческого волоса. Продолговатую форму вируса определяет слой структурного белка — матрикса. С внешней стороны матрикс окружён мембраной с поверхностными белками (гемагглютинином и нейраминидазой) и ионными каналами. Геном вируса — это восемь молекул РНК в составе спиральных комплексов. Он, как и белки, необходимые для полноценной работы вируса в зараженной клетке, находится внутри частицы.
— Гемагглютинин связывается с рецепторами на поверхности клетки, позволяя частице слиться с мембраной клеточной везикулы, проникнуть внутрь нее и доставить геном вируса в цитоплазму.
— Нейраминидаза нужна для того, чтобы только что сформированные вирионы могли отделяться от клеточной мембраны и заражать другие клетки.
— Белок матрикса — одна из основных структурных молекул вируса. Именно с этим белком связана характерная форма вирусной частицы и расположение в ней внутренних компонентов (что, в свою очередь, детерминирует правильность распаковки и процесса заражения), он же определяет её размеры. Молекулы белка матрикса связываются как с поверхностными белками, так и с нуклеопротеиновыми комплексами вируса, позволяя всем компонентам попадать в частицу.
— Белок M2 образует каналы, через которые внутрь частицы проникают ионы водорода после того, как та оказывается в клетке. Это запускает разборку вируса и высвобождение его генома.
— Нуклеопротеин упаковывает фрагменты вирусного генома в компактные спирально закрученные комплексы геномной РНК и белка NP — рибонуклеопротеиды (РНП),которые помещаются внутри вирусной частицы.
— Белок ядерного экспорта обеспечивает транспорт копий РНК генома из ядра, где они образовались, к месту сборки новых частиц вируса у поверхности зараженной клетки.
— Полимеразный комплекс необходим для создания копий РНК вируса, одна часть которых нужна для того, чтобы синтезировать структурные белки, а другая — чтобы упаковываться в новые вирусные частицы. Геном вируса несет информацию, необходимую для синтеза вирусных белков. Он представлен восемью молекулами РНК, отличающимися длиной и набором кодируемых белков.
— Мембрана вируса формируется из мембраны клетки, в которой он образовался. В ее состав входят молекулы фосфатидилхолина, фосфатидилэтаноламина, сфингомиелина и холестерина — в характерных для клеток человека пропорциях.
Процесс создания научно достоверных 3Д моделей вирусов
Пространственные структуры некоторого количества белков, входящих в состав вирионов (этадоля варьируется от 25 до 50%), описаны не полностью: проблемы возникают из-за того, что не все белковые молекулы или отдельные их фрагменты возможно кристаллизовать (а это является условием проведения рентгеноструктурного анализа). Обычно в структурах недостаёт подвижных фрагментов молекул, а также трансмембранных участков и гликозильных остатков поверхностных белков. В то же время для комплексов и ансамблей белков в большинстве случаев неизвестно, какие поверхности компонентов формируют контакты друг с другом, а какие — нет. Предсказание и описание таких взаимодействий необходимо при построении любой вируснойчастицы (для большей части входящих в неё молекулярных комплексов и структур).
Эти проблемы позволяет решить структурная биоинформатика, основанная на вычислительных методах: в данном подходе используется предсказание пространственной структуры исследуемой молекулы на основе структур схожих или родственных протеинов и исходной последовательности аминокислот, которая в большинстве случаев известна заранее. Кроме того, подобные методы позволяют рассчитать меж- и внутримолекулярные взаимодействия. В компании Visual Science эту задачу решает Отдел молекулярного моделирования и динамики: работающие в нем специалисты — молекулярные биологи и биоинформатики, обладающие научными степенями. Они проводят анализ опубликованных ранее кристаллографических данных (параллельно оценивая точность описания похожих белков), осуществляют моделирование недостающих фрагментов и в результате проводят сборку полных достоверных моделей всех компонентов и комплексов вирусной частицы, комбинируя известные данные с теми, что получены ими в Отделе. Подобный уровень сотрудничества с научным сообществом, а также столь высокая степень вовлечения в этот процесс структурных биологов из числа сотрудников Отдела молекулярного моделирования компании на данный момент недоступны ни одной студии научной и медицинской визуализации в мире — кроме Visual Science.
Дополнительные материалы: научный обзор и панорама 3Д модели вируса гриппа.
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.

Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.

Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Белки: как получают, хранят и отображают информацию об их структуре?
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.

Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.
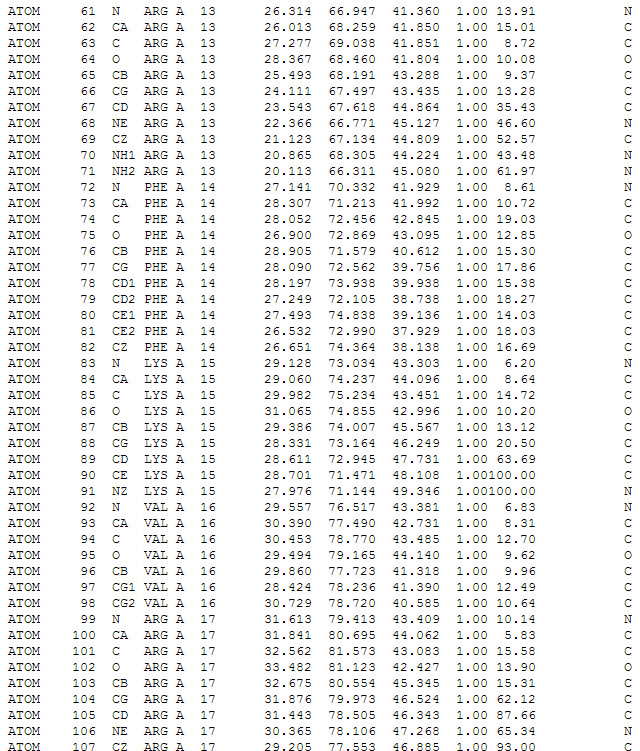
Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.

Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:

Изображения молекулы иммуноглобулина G.
Молекулярное моделирование

Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Докинг
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Посттрансляционные модификации
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Молекулярная динамика и оптимизация структур
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.

Обзор
Автор
Редакторы
Обратите внимание!
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Эволюция и происхождение вирусов
В 2007 году сотрудники биологического факультета МГУ Л. Нефедова и А. Ким описали, как мог появиться один из видов вирусов — ретровирусы. Они провели сравнительный анализ геномов дрозофилы D. melanogaster и ее эндосимбионта (микроорганизма, живущего внутри дрозофилы) — бактерии Wolbachia pipientis. Полученные данные показали, что эндогенные ретровирусы группы gypsy могли произойти от мобильных элементов генома — ретротранспозонов. Причиной этому стало появление у ретротранспозонов одного нового гена — env, — который и превратил их в вирусы. Этот ген позволяет вирусам передаваться горизонтально, от клетки к клетке и от носителя к носителю, чего ретротранспозоны делать не могли. Именно так, как показал анализ, ретровирус gypsy передался из генома дрозофилы ее симбионту — вольбахии [7]. Это открытие упомянуто здесь не случайно. Оно нам понадобится для того, чтобы понять, чем вызваны трудности борьбы с вирусами.
Из давних письменных источников, оставленных историком Фукидидом и знахарем Галеном, нам известно о первых вирусных эпидемиях, возникших в Древней Греции в 430 году до н.э. и в Риме в 166 году. Часть вирусологов предполагает, что в Риме могла произойти первая зафиксированная в источниках эпидемия оспы. Тогда от неизвестного смертоносного вируса по всей Римской империи погибло несколько миллионов человек [8]. И с того времени европейский континент уже регулярно подвергался опустошающим нашествиям всевозможных эпидемий — в первую очередь, чумы, холеры и натуральной оспы. Эпидемии внезапно приходили одна за другой вместе с перемещавшимися на дальние расстояния людьми и опустошали целые города. И так же внезапно прекращались, ничем не проявляя себя сотни лет.
Вирус натуральной оспы стал первым инфекционным носителем, который представлял действительную угрозу для человечества и от которого погибало большое количество людей. Свирепствовавшая в средние века оспа буквально выкашивала целые города, оставляя после себя огромные кладбища погибших. В 2007 году в журнале Национальной академии наук США (PNAS) вышла работа группы американских ученых — И. Дэймона и его коллег, — которым на основе геномного анализа удалось установить предположительное время возникновения вируса натуральной оспы: более 16 тысяч лет назад. Интересно, что в этой же статье ученые недоумевают по поводу своего открытия: как так случилось, что, несмотря на древний возраст вируса, эпидемии оспы не упоминаются в Библии, а также в книгах древних римлян и греков [9]?
Строение вирусов и иммунный ответ организма

Рисунок 1. Первооткрыватель вирусов Д.И. Ивановский (1864–1920) (слева) и английский врач Эдвард Дженнер (справа).

Почти все известные науке вирусы имеют свою специфическую мишень в живом организме — определенный рецептор на поверхности клетки, к которому и прикрепляется вирус. Этот вирусный механизм и предопределяет, какие именно клетки пострадают от инфекции. К примеру, вирус полиомиелита может прикрепляться лишь к нейронам и потому поражает именно их, в то время как вирусы гепатита поражают только клетки печени. Некоторые вирусы — например, вирус гриппа А-типа и риновирус — прикрепляются к рецепторам гликофорин А и ICAM-1, которые характерны для нескольких видов клеток. Вирус иммунодефицита избирает в качестве мишеней целый ряд клеток: в первую очередь, клетки иммунной системы (Т-хелперы, макрофаги), а также эозинофилы, тимоциты, дендритные клетки, астроциты и другие, несущие на своей мембране специфический рецептор СD-4 и CXCR4-корецептор [13–15].

Одновременно с этим в организме реализуется еще один, молекулярный, защитный механизм: пораженные вирусом клетки начинают производить специальные белки — интерфероны, — о которых многие слышали в связи с гриппозной инфекцией. Существует три основных вида интерферонов. Синтез интерферона-альфа (ИФ-α) стимулируют лейкоциты. Он участвует в борьбе с вирусами и обладает противоопухолевым действием. Интерферон-бета (ИФ-β) производят клетки соединительной ткани, фибробласты. Он обладает таким же действием, как и ИФ-α, только с уклоном в противоопухолевый эффект. Интерферон-гамма (ИФ-γ) синтезируют Т-клетки (Т-хелперы и (СD8+) Т-лимфоциты), что придает ему свойства иммуномодулятора, усиливающего или ослабляющего иммунитет. Как именно интерфероны борются с вирусами? Они могут, в частности, блокировать работу чужеродных нуклеиновых кислот, не давая вирусу возможности реплицироваться (размножаться).

Причины поражений в борьбе с ВИЧ
Тем не менее нельзя сказать, что ничего не делается в борьбе с ВИЧ и нет никаких подвижек в этом вопросе. Сегодня уже определены перспективные направления в исследованиях, главные из которых: использование антисмысловых молекул (антисмысловых РНК), РНК-интерференция, аптамерная и химерная технологии [12]. Но пока эти антивирусные методы — дело научных институтов, а не широкой клинической практики*. И потому более миллиона человек, по официальным данным ВОЗ, погибают ежегодно от причин, связанных с ВИЧ и СПИДом.

Подобный вирусный механизм характерен не только для ВИЧ. Он описан и при инфицировании некоторыми другими опасными вирусами: такими, как вирусы Денге и Эбола. Но при ВИЧ антителозависимое усиление инфекции сопровождается еще несколькими факторами, делая его опасным и почти неуязвимым. Так, в 1991 году американские клеточные биологи из Мэриленда (Дж. Гудсмит с коллегами), изучая иммунный ответ на ВИЧ-вакцину, обнаружили так называемый феномен антигенного импринтинга [23]. Он был описан еще в далеком 1953 году при изучении вируса гриппа. Оказалось, что иммунная система запоминает самый первый вариант вируса ВИЧ и вырабатывает к нему специфические антитела. Когда вирус видоизменяется в результате точечных мутаций, а это происходит часто и быстро, иммунная система почему-то не реагирует на эти изменения, продолжая производить антитела к самому первому варианту вируса. Именно этот феномен, как считает ряд ученых, стоит препятствием перед созданием эффективной вакцины против ВИЧ.

Открытие биологов из МГУ — Нефёдовой и Кима, — о котором упоминалось в самом начале, также говорит в пользу этой, эволюционной, версии.

Сегодня не только ВИЧ представляет опасность для человечества, хотя он, конечно, самый главный наш вирусный враг. Так сложилось, что СМИ уделяют внимание, в основном, молниеносным инфекциям, вроде атипичной пневмонии или МЕRS, которыми быстро заражается сравнительно большое количество людей (и немало гибнет). Из-за этого в тени остаются медленно текущие инфекции, которые сегодня гораздо опаснее и коварнее коронавирусов* и даже вируса Эбола. К примеру, мало кто знает о мировой эпидемии гепатита С, вирус которого был открыт в 1989 году**. А ведь по всему миру сейчас насчитывается 150 млн человек — носителей вируса гепатита С! И, по данным ВОЗ, каждый год от этой инфекции умирает 350-500 тысяч человек [33]. Для сравнения — от лихорадки Эбола в 2014-2015 гг. (на состояние по июнь 2015 г.) погибли 11 184 человека [34].
* — Коронавирусы — РНК-содержащие вирусы, поверхность которых покрыта булавовидными отростками, придающими им форму короны. Коронавирусы поражают альвеолярный эпителий (выстилку легочных альвеол), повышая проницаемость клеток, что приводит к нарушению водно-электролитного баланса и развитию пневмонии.

Рисунок 8. Электронная микрофотография воссозданного вируса H1N1, вызвавшего эпидемию в 1918 г. Рисунок с сайта phil.cdc.gov.
Почему же вдруг сложилась такая ситуация, что буквально каждый год появляются новые, всё более опасные формы вирусов? По мнению ученых, главные причины — это сомкнутость популяции, когда происходит тесный контакт людей при их большом количестве, и снижение иммунитета вследствие загрязнения среды обитания и стрессов. Научный и технический прогресс создал такие возможности и средства передвижения, что носитель опасной инфекции уже через несколько суток может добраться с одного континента на другой, преодолев тысячи километров.

Обзор
Автор
Редакторы



Спонсором приза зрительских симпатий выступила компания BioVitrum.
Мутации и вариации
Известно три разновидности вируса гриппа, опасных для человека:
- тип А (Alphainfluenzavirus) — наиболее подвержен мутациям и является постоянной головной болью Всемирной организации здравоохранения (ВОЗ);
- тип В (Betainfluenzavirus) — более стабилен, но все же может видоизменяться;
- тип С (Gammainfluenzavirus) — наиболее стабилен, поэтому к нему вырабатывается длительный иммунитет. Эпидемичных вспышек не дает, чаще всего приводит к нетяжелому заболеванию у детей.
Если бы все типы вируса гриппа были похожи на тип С, больших проблем с ними не было. Однако тип А постоянно мутирует, поэтому довольно часто появляются его новые вариации (штаммы), с которыми наша иммунная система еще не знакома [2]. Из-за этой изменчивости классификация вирусов гриппа достаточно сложная: внутри каждого типа существуют подтипы (в случае с типом В — линии), в которые объединяют штаммы вируса. Причем, штаммы подтипов могут быть как родственными (то есть эволюционно недалеко ушедшими друг от друга), так и непохожими.
Кому опасен грипп?
Причем в случае с беременными женщинами риск касается не только будущей мамы, но и ее ребенка: грипп во время беременности более чем в 7 раз повышает риск госпитализации, а также может привести к преждевременным родам (около 30% случаев), мертворождению и малому весу при рождении [2], [6]. Поэтому во многих странах мира (США, Великобритания, Австралия, Италия) беременным рекомендована вакцинация против гриппа. Делают это по двум причинам:

Рисунок 1. Строение вируса гриппа (типы А и В)
Когда лучше сделать прививку?
Вакцинация против гриппа — это ежегодная прививка , которая защищает от трех или четырех наиболее распространенных в данной местности штаммов вируса. Это значит, что каждый год на основании рекомендаций ВОЗ и региональной ситуации национальные комитеты по контролю над гриппом составляют рекомендации антигенного состава будущей вакцины [15], [16]. Однако чаще всего эти рекомендации совпадают с рекомендациями ВОЗ, которые публикуются отдельно для северного и южного полушарий.
Большинству людей прививают одну дозу вакцины, однако детям от шести месяцев до двух лет (и до девяти лет в случае их первой вакцинации [17]) рекомендованы две дозы с минимальным интервалом в один месяц. Исследования показывают, что в этом случае эффективность вакцинации увеличивается [18], [2].
Состав противогриппозных вакцин все время меняется: например, в сезоне 2019–2020 были заменены оба штамма вируса типа А, и в итоге в четырехкомпонентную вакцину вошли:
- A/Brisbane/02/2018 (H1N1);
- A/Kansas/14/2017 (H3N2);
- B/Colorado/06/2017 (линия B/Victoria/2/87);
- B/Phuket/3073/2013 (линия B/Yamagata/16/88).
В трехкомпонентную вакцину, соответственно, рекомендовано включить первые три штамма вируса [16]. Однако бывает и так, что каждый год в составе вакцин повторяется название одного из штаммов. Значит ли это, что постоянно прививают одно и то же? Нет, даже в этом случае штаммы могут существенно различаться, в том числе и по генам, не входящим в классификацию.
Как долго длится иммунитет после вакцинации и имеет ли он пролонгированный эффект на будущий год? К сожалению, эффективность прививок против гриппа недолговечна. Она зависит от времени, прошедшего с момента прививки и штамма вируса: в среднем, считается, что защита снижается примерно на 7% в месяц для H3N2 и штаммов линии В и на 6–11% — для H1N1 [17]. Конечно, скорость и степень снижения могут различаться, но эффективной защиты, скорее всего, хватает на год [11].
Как выбирают штаммы и почему четыре лучше трех?
В течение всего года специалисты NICs анализируют циркулирующие штаммы вирусов на основании лабораторных анализов пациентов с респираторными заболеваниями, выделяют из общей массы пробы с вирусом гриппа и выбирают подходящих кандидатов для дальнейшего изучения в одном из пяти центров ВОЗ (WHO CCs) [19]. Отбор идет по принципу типичности вируса для данного региона и новизне, которую определяют по его реакции с антителами из набора ВОЗ. Дальнейшая работа осуществляется уже в центрах ВОЗ, где штаммы культивируют, анализируют, сравнивают между собой, составляют карты антигенности, строят математические модели и в итоге на основании всех этих данных выбирают претендентов в состав вакцины [19]. Как происходит этот процесс и сколько времени занимает каждая стадия, показано на рисунке 2.

Рисунок 2. Процесс отбора штаммов для противогриппозной вакцины
И наконец, дважды в год проходят Сезонные совещания ВОЗ, посвященные составам противогриппозных вакцин (Seasonal influenza vaccine composition meeting), на которых объявляют рекомендации для будущего сезона: в феврале — для северного полушария, в сентябре — для южного. Как только составы обнародованы, и производители получают вакцинные штаммы, запускается процесс производства, на который уходит около полугода (видео 1). Однако ошибки в планировании могут задержать весь цикл, что скажется на количестве произведенной вакцины или на сроках ее поставки.
Видео 1. Производство противогриппозных вакцин
Почему все-таки четырехкомпонентная вакцина лучше трехкомпонентной, если циркулирующих штаммов гораздо больше? Все дело в линии В, вирусы которой обычно циркулируют вместе, но в разных пропорциях [3], поэтому в случае с вакцинами, состоящими из трех компонентов, штамм линии В всегда является компромиссным вариантом. Экспертам ВОЗ приходится выбирать большее из двух зол, но так как невозможно точно предсказать ситуацию, которая будет наблюдаться через восемь месяцев, периодически случаются ошибки, сказывающиеся на эффективности вакцины. Например, в сезоне 2017–2018 она оказалась ниже ожидаемой, так как ВОЗ прогадала со штаммом вируса типа В, предположив, что доминировать будет линия Victoria, а оказалось — Yamagata [20]. Кроме того, уже не первый год наблюдается низкая эффективность вакцины в отношении штамма H3N2. Точная причина неизвестна, но существует несколько предположений:
- Адаптация штамма во время производства может приводить к некоторым изменениям (антигенному несоответствию), и иммунитет развивается уже к новому штамму, который отличается от циркулирующего.
- Циркулирующие штаммы подтипа H3N2 меняются быстрее, чем другие — им хватает полугода (то есть времени, прошедшего с момента объявления рекомендаций ВОЗ), чтобы измениться и стать менее похожим на вакцинный штамм.
- Стандартной дозы, содержащейся в вакцине, может быть недостаточно для эффективной защиты [18], [21].
Какой должна быть идеальная вакцина?
Вакцины против гриппа бывают живыми (интраназальные вакцины, применяются редко) и инактивированными. Современные инактивированные делятся на нескольких категорий:

Рисунок 3. Виды антигенов инактивированных вакцин. а — Инактивированный вирусный вирион в цельновирионной вакцине. б — Расщепленный инактивированный вирион в сплит-вакцине. в — Частички антигена в субъединичной вакцине.
Все вышеперечисленные вакцины являются вакцинами против сезонного гриппа .
В отдельную группу выделяют препандемические и пандемические вакцины. Их производят в случае возникновения угрозы пандемии. Препандемические (зоонозные) состоят из штамма зарождающегося вируса животного происхождения, который, по мнению экспертов, обладает пандемическим потенциалом, пандемические — из штамма, вызвавшего пандемию (такие вакцины появляются на волне заболеваемости) [15].
Однако выбрать штаммы для состава — лишь полдела. Главное, чтобы вакцина была эффективной. Для этого существуют определенные критерии.
Во-вторых, существуют требования к титрам антител после вакцинации (в том числе и для вакцин с адъювантами), которые указаны в таблице 1.
| Показатель | Люди от 18 до 60 лет | Люди старше 60 лет |
|---|---|---|
| 1. Кратность нарастания среднего геометрического титра антител после вакцинации (GMT increase) | 2,5 раза | 2 раза |
| 2. Уровень сероконверсии * (процент привитых с нарастанием титра антител минимум в четыре раза по сравнению с исходым) | 40% | 30% |
| 3. Уровень серопротекции (число лиц с защитным титром) ** | 70% | 60% |
| * — В тестах, измеряющих ингибирование гемагглютинина (HI), сероконверсия соответствует отрицательной сыворотке до вакцинации (HI < 1:10) и сыворотке крови после вакцинации HI ≥ 1:40. ** — Серопротекция соответствует проценту привитых с сывороткой HI ≥ 1:40. | ||
Для сезонных вакцин необязательно соблюдение всех трех условий; соответствие всем требованиям необходимо только для пандемических [24]. Мало того, сейчас титр HI ≥ 1:40 уже не считается надежным фактором для определения эффективности защиты (50–70% против клинических симптомов гриппа), так как уровни защиты могут варьировать в зависимости от индивидуальных характеристик, групп населения, возрастных групп и даже от типа вакцины [25].
В-третьих, есть отдельные требования к вакцинам, содержащим адъюванты:
- Совместимость адъюванта с антигенными компонентами вакцины.
- Доказательство последовательной связи адъюванта с вакцинными антигенами во время производства и в течение срока годности.
- Данные о влиянии адъюванта на эффективность вакцины.
- Биохимическая чистота адъюванта [23].
Если все это суммировать, то идеальная вакцина должна быть безопасной (низкореактогенной ), содержать 15 мкг гемагглютинина на дозу, вызывать определенные уровни титров антител у привитых в зависимости от их возраста (при этом количество эффективно привитых должно быть не менее 70% среди взрослого населения до 60 лет). Если же вакцина содержит адъювант, он должен быть безопасным, связанным с антигенами и вызывать иммунный ответ в соответствии со строгими стандартами.
Что касается безопасности, то благодаря широкому использованию сплит- и субъединичных вакцин, прививки против гриппа демонстрируют низкую реактогенность. В основном наблюдаются местные реакции (у 10–64 привитых из 100) и повышение температуры (чаще всего у детей: 12 из 100 привитых) [26].
Вакцинация против гриппа и аллергия на куриный белок
В противопоказаниях к вакцинам против гриппа указано, что их нельзя прививать людям, у которых есть аллергические реакции на любой из компонентов, в том числе и на белок куриного яйца [27]. Однако в международной практике людей с аллергией на куриный белок совершенно спокойно прививают как против гриппа, так и против кори, краснухи и паротита, хотя вирусы для этих вакцин выращивают с использованием куриных эмбрионов. Вакцинации аллергиков дали зеленый свет после серии исследований [28–30], в которых изучали реактогенность у людей с аллергическими реакциями на куриный белок: в итоге эти вакцины признали безопасными, и теперь прививают даже людям с анафилактической реакцией на куриный белок (единственное, таких пациентов нельзя прививать в аптеках или школах, как это делают в некоторых странах — только в медицинских центрах, где есть противошоковые медикаменты).
Во время производства вакцины клеточную культуру подвергают сериям центрифугирований и ультрафильтраций, которые позволяют отделить вирусные частицы от остальных белков. Конечно, эта технология не идеальна, но даже если в препарат вдруг что-то и попадает, то лишь следовые количества овальбумина — основного белка куриного яйца: ≤ 1 мкг на 0,5 мл дозы инактивированной и 0,24 мкг на 0,2 мл дозы живой вакцины [31]. Поэтому основным противопоказанием для вакцинации против гриппа являются только тяжелые реакции на введение этих вакцин в прошлом (реакция на предыдущую дозу и аллергия на куриный белок не всегда связаны между собой: человек мог отреагировать на другой компонент, например, на неомицин) [27], [31].
Чем же прививаться?
Это вопрос, который волнует многих. В России прививают следующими вакцинами:
В какие противогриппозные вакцины добавляют адъюванты?
Муки выбора
Но, честно говоря, таких исследований единицы, поэтому выводы приходится делать по косвенным данным — официальной статистике заболеваемости гриппом в зависимости от количества привитых в нашей стране (рис. 4).

Рисунок 4. Заболеваемость гриппом и количество привитых против гриппа в России за 1996–2018 годы
Автор благодарит врача-биофизика Кирилла Скрипкина за помощь в подготовке материала.
Читайте также: