
Болезнь крона и аутоиммунный гепатит
Обновлено: 18.04.2024
В настоящее время аутоимунный гепатит (АИГ) определяется как "персистирующее (неразрешившееся) воспаление печени неизвестной этиологии, характеризующееся преимущественно перипортальным гепатитом или более обширным воспалительным процессом, сопровождающееся гипергаммаглобулинемией, присутствием тканевых аутоантител в сыворотке и в большинстве случаев отвечающее на иммуносупрессивную терапию".
АИГ является частью так называемого "синдрома хронического гепатита", который характеризуется устойчивым гепатоцеллюлярным воспалением в течение последних 6 месяцев и подъемом трансаминаз более чем в 1,5 раза выше верхнего предела нормы (см. рубрику "Хронический гепатит, не классифицированный в других рубриках" - K73.-). В редакциях МКБ-10 до сентября 2013 г. АИГ кодировался как "Хронический активный гепатит, не классифицированный в других рубриках" - K73.2.
Примечание 1
Первое описание хронического гепатита с "сосудистыми звездочками", высокими показателями СОЭ, гипергаммаглобулинемией, аменореей и хорошим эффектом от лечения кортикотропином у 6 молодых женщин относится к 1950 году (Waldenstrom). В последующем была отмечена ассоциация данного варианта хронического гепатита с различными аутоиммунными синдромами и наличием антинуклеарных антител в сыворотке, в связи с чем появился термин "люпоидный гепатит" (Mackay, 1956). В 1965 году был предложен термин "аутоиммунный гепатит".
Систематизированные исследования клеточной и молекулярной иммунопатологии, клинических симптомов и лабораторных показателей впоследствии привели к выделению АИГ как отдельной нозологической единицы, являющейся серологически гетерогенным заболеванием, которое требует определенной терапевтической стратегии (Strassburg, 2000). Исследования последних лет показали, что АИГ представляет гетерогенную группу заболеваний, отличающихся по антигенам и соответствующим циркулирующим в сыворотке антителам (см. раздел "Классификация").
- "Печеночная недостаточность, не классифицированная в других рубриках (острые и подострые поражения печени)" - K72.-
Классификация
Вопросы классификации аутоиммунного гепатита (АИГ) остаются спорными в связи с разнородностью выявляемых иммунологических изменений.
I. Традиционно выделялось два типа АИГ (тип I и тип II). В настоящее время дополнительно выделяется тип III, что признается не всеми авторами.
В основу общепринятой классификации АИГ положен спектр выявляемых аутоантител:
АИГ типа I (АИГ-1) характеризуется циркуляцией антинуклеарных антител (ANA) у 70-80% больных и/или антигладкомышечных аутоантител (SMA) у 50-70% больных , нередко в сочетании с антинейтрофильными цитоплазматическими антителами р-типа (p-ANCA).
АИГ-1 может развиваться в любом возрасте, но более типичен в 10-20 лет и в период постменопаузы. Формирование цирроза отмечают у 43% нелеченых больных в течение первых 3 лет. У большинства пациентов наблюдают хороший ответ на терапию ГКС , при этом у 20% сохраняется стойкая ремиссия после отмены иммуносупрессоров.
АИГ типа II (АИГ-2) характеризуется циркуляцией антител к микросомам печени и почек 1-го типа (анти-LKM-l), определяемых у 100% больных, иногда в сочетании с анти-LКМ-3 и антителами к печеночному цитозольному протеину (анти-LC-1).
АИГ-2 встречают существенно реже (10-15% больных АИГ) и преимущественно у детей от 2 до 14 лет. Взрослые составляют 20% от общего числа пациенток в Европе и лишь 4% в США.
Течение заболевания характеризуется более высокий биохимической и гистологической активностью. Цирроз за 3-летний период формируется в 2 раза чаще (у 82%), чем при АИГ-1 , что определяет худший прогноз. При АИГ-2 наблюдают более выраженную резистентность к медикаментозной иммуносупрессии; отмена препаратов обычно ведет к рецидиву.
АИГ типа III (АИГ-3) характеризуется наличием в крови антител к растворимому печеночному антигену (анти-SLA) и печеночно-панкреатическому антигену (анти-LP). Этот тип выделяют не все авторы; многие рассматривают его как подтип АИГ-1, учитывая одинаковое клиническое течение и частое (74%) выявление соответствующих серологических маркёров (ANA и SMA).
II. В последние годы спектр классификации АИГ дополнен следующими формами:
- "криптогенный хронический гепатит" (около 20% пациентов) при наличии диагностических критериев АИГ-1, но в отсутствие аутоантител (так называемый "4-й тип АИГ);
- вариантными формами аутоиммунного гепатита (перекрестные синдромы).
Хотя споры по поводу уточнения форм, перекрывающихся с другими аутоиммунными заболеваниями печени, ведутся, следующие ниже вариантные формы АИГ считаются наиболее общепризнанными.
1. Синдром перекрытия АИГ и первичного билиарного цирроза (см. "Первичный билиарный цирроз" - K74.3):
1.1. Гистологические признаки АИГ положительны и одновременно серологический диагноз первичного билиарного цирроза (антимитохондриальные антитела (АМА)) также положительный.
1.2. Присутствуют гистологические признаки первичного билиарного цирроза и серологические результаты АИГ (ANA или SMA-положительные, AMA-отрицательные). Данная форма иногда считается аутоиммунным холангитом или АМА-негативным первичным билиарным циррозом.
2. Синдром перекрытия АИГ и первичного склерозирующего холангита (см. "Холангит" - K83.0): имеются серологические признаки АИГ, но гистологические результаты и нарушения, выявленные при холангиографии, характерны для первичного склерозирующего холангита.
Этиология и патогенез
Этиология аутоиммунного гепатита (АИГ) неясна.
Существует множество гипотез, пытающихся объяснить возникновение АИГ. Однако разнообразная иммунологическая картина и вариабельность ассоциированной патологии затрудняют задачу.
Вероятно, АИГ возникает вследствие сложного взаимодействия следующих факторов:
1. Генетическая предрасположенность. Гены HLA главного комплекса гистосовместимости (MHC), расположенные на коротком плече хромосомы 6, по-видимому, играют основную роль в предрасположенности к болезни. Существует также доказательство роли других, не HLA локусов, которые кодируют факторы комплемента, иммуноглобулинов и Т-клеточных рецепторов.
2. Триггеры.
В качестве возможных запускающих факторов АИГ, помимо HAV, HBV, HCV, EBV, рассматриваются также вирус простого герпеса (HSV1), цитомегаловирус (CMV) и вирус кори. При этом обращается внимание на то, что некоторые из них (в частности HAV, HCV, EBV и вирус кори) могут в течение ряда лет персистировать "незамеченными" в лимфоцитах периферической крови.
Немаловажная роль отводится лекарствам (например, оксифенизатин, миноциклин, тикринафен, дигидралазин, метилдопа, нитрофурантоин, диклофенак, атровастатин, интерферон, пемолин, инфликсимаб, эзетимиб) и некоторым травам, применяемым в народной медицине.
3. Аутоантигены. Наиболее часто рассматриваемые:
- асиалогликопротеиновый рецептор (ASGP-R) для антител против ASGP-R;
- цитохром P450 2D6 (CYP2D6) для anti-LKM-1 аутоантител.
4. Дисфункция иммунорегуляторных механизмов. АИГ может развиться как компонент синдрома аутоиммунной полиэндокринопатической эктодермальной дистрофии (APECED) у 10-20% пациентов. При этом достаточно часто в процесс вовлекается не только печень, но и крупные железы внешней и внутренней секреции, в том числе поджелудочная железа, щитовидная железа, слюнные железы.
Патогенез
Главный патогенетический механизм развития повреждения печени - потеря иммунной толерантности к собственным тканям, что обусловливает прогрессирование некро-воспаления и фиброза в печени и отражает сложное взаимоотношение между запускающими аутоиммунный процесс факторами, аутоантигенами, генетической предрасположенностью и иммунорегуляторными процессами.
Гистологические изменения, отмечающиеся при АИГ, не являются патогномоничными, однако достаточно типичны.
Наблюдается круглоклеточная инфильтрация портальных полей различной плотности (преимущественно Т-лимфоцитами). Воспалительные инфильтраты не захватывают желчные протоки или сосудистую систему, но могут проникать через основную пластинку к печеночной дольке, вызывая отшнуровку и разрушение отдельных гепатоцитов или их небольших групп (ступенчатые некрозы, часто обозначаемые как пограничный гепатит (interface hepatitis)).
В случае когда дорожки некроза соединяются с подобными участками соседних перипортальных полей, говорят о мостовидных некрозах. Они могут распространяться вплоть до центральных участков печеночной дольки.
Таким образом, АИГ характеризуется соседством перипортального и лобулярного гепатита.
В далеко зашедших стадиях очаги некроза замещаются соединительной тканью и развивается цирроз с островками паренхимы и узлами-регенератами различных размеров. Изменения желчных протоков, гранулёмы, скопления железа и меди при этом отсутствуют.
Поражение желчных протоков ранее считалось возможным признаком гистологической картины АИГ-1. В настоящее время оно исключает данный диагноз и указывает на билиарный цирроз печени. Все вышесказанное относится и к отложению меди, наблюдающемуся при всех формах холестаза и свидетельствующему о холестатическом заболевании (билиарный цирроз печени, первичный склерозирующий холангит) или перекрестном синдроме, но не об АИГ (определение Международной группы по изучению аутоиммунного гепатита, IAIHG).
Эпидемиология
Возраст: кроме младенцев
Признак распространенности: Редко
Соотношение полов(м/ж): 0.27
Частота. 1-1,9 случаев на 100 000 населения европеоидной расы в США и Европе.
Есть мнение, что заболеваемость существенно ниже в странах Азии и Африки, в связи с превалированием там вирусных гепатитов и генетических особенностей, присущих европеоидной расе.
Пол. Женщины страдают чаще, чем мужчины.
АИГ-1 является наиболее распространенной формой АИГ, и 78% больных составляют женщины (соотношение женщин и мужчин составляет - 3,6:1).
Около 95% больных АИГ-2 составляют женщины.
В среднем соотношение полов женщины/мужчины оценивается как 4:1.
Возраст. Заболеваемость АИГ характеризуется возрастной бимодальностью, то есть двумя пиками.
АИГ может развиться в любой возрастной группе, но АИГ-1 чаще всего затрагивает людей в возрасте от 10 до 30 лет и от 40 до 60 лет.
АИГ-2 поражает главным образом детей в возрасте от 2 до 15 лет.
Таким образом, наибольшая заболеваемость встречается в группе молодых девушек и женщин европеоидной расы. Тем не менее, АИГ может возникнуть у людей любого возраста, включая младенцев и пожилых людей. Не следует упускать из виду у лиц старше 70 лет, причем мужчины в этом возрасте могут быть затронуты чаще, чем женщины.
Соотношение заболеваемости АИГ-1 к АИГ-2 оценивается как 1.5-2:1 в Европе и Канаде и 6-7:1 в Северной Америке, Южной Америке и Японии.
Факторы и группы риска
Основные факторы риска:
- женский пол;
- генетическая предрасположенность;
- нарушения регуляции иммунного ответа.
Клиническая картина
Клинические критерии диагностики
недомогание, снижение аппетита, дискомфорт в животе, гепатомегалия, желтуха, энцефалопатия, кожный зуд, артралгия мелких суставов, тошнота, лихорадка, телеангиоэктазии, спленомегалия, асцит, диарея, миалгии, гирсутизм, аменорея, кровоточивость
Cимптомы, течение
I. Аутоиммунный гепатит (АИГ) может проявляться как острый или хронический гепатит, а также как сформировавшийся (и/или прогрессирующий) цирроз печени Цирроз печени - хроническая прогрессирующая болезнь, характеризующаяся дистрофией и некрозом печеночной паренхимы, сопровождающимися ее узловой регенерацией, диффузным разрастанием соединительной ткани и глубокой перестройкой архитектоники печени.
Подробно .
Примерно у трети пациентов имеется симптоматика острого гепатита, характеризующаяся лихорадкой, болями в печени и желтухой.
В редких случаях АИГ протекает как молниеносная печеночная недостаточность (см. "Острая и подострая печеночная недостаточность" - K72.0).
Цирроз печени выявляется изначально у 20% пациентов с АИГ и еще у 20-40% гепатит в дальнейшем прогрессирует в цирроз.
Наиболее часто начало заболевания бессимптомное или характеризуется очень неспецифичными жалобами, либо жалобами, связанными с ассоциированной аутоиммунной патологией.
II. Общие, но неспецифические, симптомы включают в себя следующие:
При исследовании детей с АИГ зафиксированы следующие основные клинические признаки:
- анорексия Анорексия — синдром, заключающийся в отсутствии аппетита, чувства голода, либо в сознательном отказе от пищи
(47%);
III. Параллельно с АИГ у 38% взрослых пациентов могут выявляться внепеченочные иммунные заболевания, в том числе:
Что такое цирроз печени? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, гастроэнтеролога со стажем в 15 лет.
Над статьей доктора Васильева Романа Владимировича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов

Определение болезни. Причины заболевания
Цирроз печени (ЦП) — это хроническое дегенеративное заболевание печени, связанное с диффузным патологическим процессом, при котором нормальные клетки печени повреждаются, а затем замещаются рубцовой тканью, образуя избыточный фиброз и структурно-анатомические регенераторные узлы.
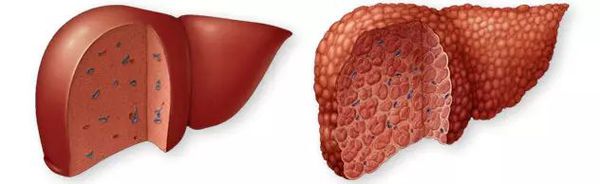
Этиология
По этиологическим характеристикам можно выделить:
- распространённые формы ЦП;
- редкие формы ЦП.
К распространённым относят вирусные (В, С, D), алкогольные и метаболические формы цирроза печени.
Редкими формами ЦП являются:
- аутоиммунные, лекарственные, токсические, первичные и вторичные билиарные циррозы;
- генетически обусловленные патологии — гемохроматоз (нарушение обмена железа), болезнь Вильсона — Коновалова, дефицит белка альфа-1-антитрипсина, гликогеноз IV типа (недостаток ферментов), галактоземия, наследственная тирозинемия и непереносимость фруктозы;
- нарушение венозного оттока из печени — венокклюзионные формы ЦП (болезнь Бадда — Киари);
- тяжёлая правожелудочковая сердечная недостаточность;
- флебопортальные циррозы (типа Банти).
Пути заражения
Заразиться циррозом печени нельзя. Однако, если он вызван вирусным гепатитом, то возбудитель может передаться через кровь, при половых контактах и от матери к ребёнку.
Основную роль в возникновении и развитии вирусного ЦП играют симптомные, малосимптомные и бессимптомные формы острого вирусного гепатита В, С, а также одновременное заболевание гепатитами В и D с последующим переходом в активный хронический вирусный гепатит. У большинства больных интервал между острым гепатитом С и клинически выраженными проявлениями ЦП превышает 30 лет. Только у мужчин, употребляющих более 50 г спирта в день, выраженные формы ЦП возникают через 13-15 лет.
Наиболее частыми причинами смерти больных ЦП является:
- большая печёночная недостаточность;
- кровотечение из варикозно расширенных вен пищевода;
- первичный рак печени;
- иммунопротективная недостаточность, влекущая за собой активизацию инфекционных (микробных) процессов, в первую очередь спонтанного бактериального перитонита и пневмонии, а также возникновение оксидативного стресса.
У больных в терминальной (заключительной) фазе заболеваний печени в основном наблюдаются декомпенсированные формы цирроза печени: асцит, варикозное расширение вен пищевода и желудка, энцефалопатия и желтуха.
Особенности цирроза печени у детей
Заболевание у детей встречается крайне редко и обычно связано:
- с аутоиммунным поражением печени;
- кардиогенными заболеваниями — лёгочной гипертензией и хронической сердечной недостаточностью;
- болезнью Бадда — Киари;
- врождёнными болезнями накопления — наследственным гемохроматозом, лизосомальными болезнями накопления, болезнью Вильсона — Коновалова;
- флебопортальным циррозом (типа Банти).
Прогноз у таких детей неблагоприятный, чаще всего они погибают, так как не успевают попасть к гепатологу и выяснить диагноз. Также они обычно страдают от множества сопутствующих болезней, в том числе от основного заболевания, ставшего причиной цирроза.
Проявления заболевания у детей и взрослых схожи. Единственный эффективный метод лечения цирроза у детей — это пересадка печени. Поэтому крайне важно вовремя диагностировать заболевание и встать в очередь на пересадку печени.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы цирроза печени
Цирроз печени в течение длительного времени может протекать латентно, т. е. бессимптомно.
Клиническая картина ЦП зависит от его формы и течения, активности основного заболевания, а также наличия или отсутствия печёночно-клеточной недостаточности, синдрома портальной гипертензии, холестаза и внепечёночных проявлений.
Основные общие симптомы, которые чаще всего встречаются при ЦП:
- повышенная утомляемость;
- похудение;
- нарушения сознания и поведения;
- ухудшение аппетита и чувство дискомфорта в животе;
- пожелтение кожи, белковых оболочек глаз и слизистой;
- осветление или обесцвечивание кала;
- потемнение мочи;
- болевые ощущения в животе;
- отёки;
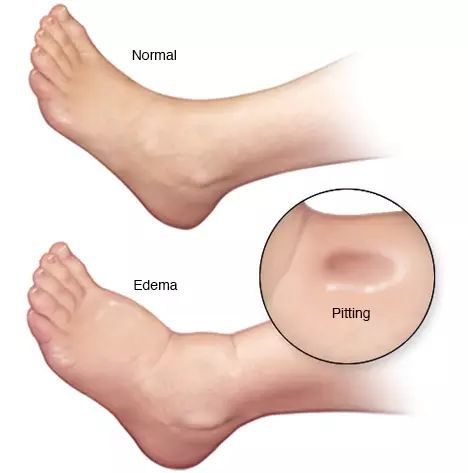
- асцит (скопление жидкости в брюшной полости);
- кровотечения из носа, желудочно-кишечного тракта, дёсен или геморроидальных узлов, а также подкожные кровоизлияния;
- часто возникающие бактериальные инфекции (например, органов дыхания);
- снижение полового влечения;
- кожный зуд.
Симптомы распространённых форм ЦП
При высокоактивном ЦП, кроме общей утомляемости, осветления стула и потемнения мочи, может возникать тупая боль в правом подреберье и вздутие живота.
Во время осмотра часто выявляют:
- субиктеричность (желтушность) склер;
- расширение вен брюшной стенки, напоминающее голову медузы;
- венозный шум при выслушивании в эпигастральной области живота (шум Крювелье — Баумгартена);
- серо-коричневатый цвет шеи;
- гинекомастию (увеличение грудных желёз);
- гипогонадизм (у мужчин);
- контрактуру Дюпюитрена (укорочение сухожилий ладоней).
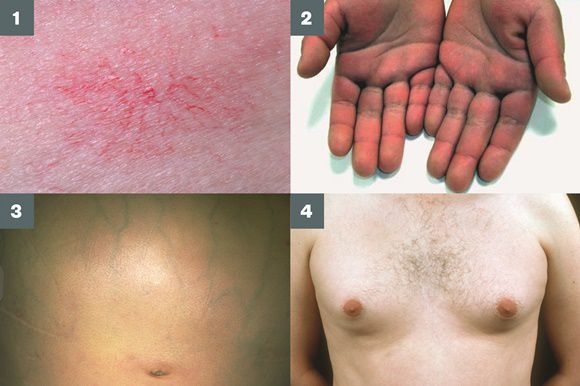
Три последних признака цирроза печени зачастую наблюдаются при алкогольных ЦП.
В области грудной клетки в 50-80% случаев наблюдаются телеангиэктазии кожи (расширения мелких сосудов), чаще при алкогольных ЦП. Пальпаторно печень отчётлива уплотнена, имеет неровный нижний край. Размеры печени различны — от значительного увеличения до уменьшения.
Часто при пальпации выявляется умеренно увеличенная селезёнка, причём её край может выступать из-под рёберной дуги на 1-3 см.
При развитии ЦП появляются симптомы белково-энергетической недостаточности, асцит, отёки, а также печёночный запах при тяжёлой печёночной недостаточности.
Симптомы при малоактивных и начальных стадиях ЦП
Данные формы ЦП зачастую протекают бессимптомно и выявляются в ходе периодических медицинских осмотров, диспансеризации, а также как случайная находка при обследовании пациента со смежной патологией или внепечёночными проявлениями.
При малоактивном ЦП, как правило, не возникают жалобы, связанные с печенью. Во время активного расспроса можно выявить весеннее снижение работоспособности, частые болезни, после которых возможны кровоточивость дёсен и потемнение мочи. Такие пациенты хуже, чем раньше, переносят длительные физические и нервно-психические нагрузки.
Желтухи и выраженного увеличения билирубина, за исключением периода интеркурентного острого гепатита, нет. Неяркая телеангиоэктазия кожи (сосудистые звёздочки) в области грудной клетки наблюдаются у 40-60% людей с ЦП.
Телеангиоэктазии кожи, плотная печень с фестончатым краем и умеренно увеличенная селезёнка — ценная клинико-диагностическая триада, которая с вероятностью 80-90% свидетельствует о ЦП или далеко зашедшем активном хроническом гепатите.
Патогенез цирроза печени
В основе патофизиологии цирроза лежит повреждение и некроз паренхимы (основной ткани) печени с деструкцией и гибелью гепатоцитов (клеток печени), а также системное поражение интерстициальной ткани.
При всех формах ЦП нарушается иммунологическое равновесие организма, преобладающими становятся аутоиммунные процессы: иммунная система человека принимает собственные клетки печени за чужеродные и повреждает их. В конечном итоге, это приводит к разрушению гепатоцитов и структуры печени в целом. Однако при этом каждая форма ЦП имеет свои патогенетические особенности:
- при вирусных гепатитах повреждающим агентом является сама вирусная частица, которая, размножаясь в клетке, разрушает её, вызывая цитолиз;
- при алкогольном ЦП прямое токсическое воздействие на мембраны гепатоцитов оказывает ацетальдегид с развитием алкогольной жировой болезни печени и алкогольного стеатогепатита;
- при метаболическом ЦП ведущую роль в патогенезе играет ожирение и сахарный диабет через стадию неалкогольного стеатогепатита с инсулинорезистентностью и последующей запрограммированной гибелью клеток печени.
В основе патогенеза более редких причин цирроза печени лежат ещё более частные механизмы развития повреждения и разрушения гепатоцитов и структуры печени:
- нарушение обмена и накопления железа при гемохроматозе;
- накопление меди при болезни Вильсона — Коновалова;
- окклюзия в системе воротной вены при гепатопортальном склерозе.
Цирроз формируется на протяжении многих лет. С течением времени происходят изменения генетического аппарата клеток печени, в результате чего появляются новые патологические клетки. Этот процесс в печени является иммуновоспалительным, он поддерживается чужеродными агентами, в роли которых могут выступать разные субстраты:
- вирус гепатита В;
- алкогольный гиалин;
- денатурированные белки;
- некоторые лекарственные средства;
- медьбелковые и железобелковые комплексы (ферритин).
В итоге повреждения паренхимы печени развивается гепатоцеллюлярная (печёночно-клеточная) недостаточность за счёт диффузного фиброза и трансформации ткани печени в анормальные узлы-регенераты. [3] [4] [5]
Классификация и стадии развития цирроза печени
В 1974 году на съезде гепатологов в Акапулько (Мексика) была принята единая морфологическая классификация, которую позже уточнили и несколько доработали эксперты ВОЗ. В настоящее время она является общепринятой.
Патогенез АГ сложен. Полагают, что это ответ генетически предрасположенного организма на какой-то внешний агент, который является пусковым моментом в развитии аутоиммунных процессов, вызывающих прогрессирующие воспалительно-некротические изменени
 |
Патогенез АГ сложен. Полагают, что это ответ генетически предрасположенного организма на какой-то внешний агент, который является пусковым моментом в развитии аутоиммунных процессов, вызывающих прогрессирующие воспалительно-некротические изменения, приводящие к фиброзу и циррозу печени (ЦП). Генетически детерминированная предрасположенность к этому заболеванию выявлена во многих исследованиях. Доказано, что большая часть больных АГ имеют фенотип по антигенам главного комплекса гистосовместимости HLA-B8, HLA-DR4, DR3 и DR52a. Пусковой агент пока неизвестен, однако есть некоторые данные о роли вирусов гепатита [31, 26], кори [27], Эпштейн-Барр вируса [32], а также интерферона (ИФН) [14] как инициаторов начала АГ.
| Аутоиммунный гепатит (АГ) — хроническое воспалительное заболевание печени невыясненой этиологии, характеризующееся определенными лабораторными, клиническими и гистологическими признаками. Болеют им в основном женщины молодого возраста |
АГ — это прогрессирующее воспаление печени, характеризующееся наличием некрозов в перипортальной, септальной зонах (ступенчатые некрозы) или, более широко, лобулярным гепатитом (ЛГ), гипергаммаглобулинемией и аутоантителами в сыворотке крови [7]. Портальные тракты печени на биоптатах находят расширенными с накоплением в них обширных инфильтратов, имеющих разный клеточный состав: лимфоциты, макрофаги, плазматические клетки. ЛГ — дольковый гепатит, когда некрозы выявляются во второй и третьей зонах ацинусов, а также обнаруживается внутридольковая лимфоидноклеточная инфильтрация, которая выражена значительно больше, чем инфильтрация портальных трактов. ЛГ является частью гистологической картины АГ, если он выявляется одновременно с перипортальным гепатитом. По гистологической картине на АГ может указывать, кроме вышеперечисленного, наличие многоядерных гепатоцитов [2].
Наконец, картина фиброза может присутствовать в той или иной степени даже при умеренной степени активности АГ, а в запущенных случаях, особенно при отсутствии эффективной терапии, формируются мостовидные некрозы и, в конце концов, ЦП.
Хотя гистологическая картина при АГ очень характерна, все-таки она неспецифична. Отличительной чертой АГ является обнаружение в биоптатах преимущественно плазматических клеток, так как выраженная инфильтрация в портальной, перипортальной зоне, вовлечение в процесс долек печени — в равной мере присущи и хроническому вирусному гепатиту (ХВГ).
Одной из основных клинических характеристик АГ является обнаружение аутоантител к клеточным и субклеточным структурам клеток разных органов [22]. Типичным маркером АГ являются антитела к ядрам клеток — ANA. Из других маркеров выявляются антитела к клеткам гладкой мускулатуры (SMA), антитела к микросомам клеток печени и эпителиальных клеток клубочкового аппарата почек (LKM), антитела к растворимому печеночному антигену (SLA), антитела к антигенам (цитокератины 8, 18) мембран гепатоцитов — LMA.
Клинические проявления АГ очень разнообразны [1, 3, 4]. С одной стороны, встречаются бессимптомные формы, когда случайно выявляется повышение АЛТ, АСТ, а с другой — острое начало болезни с тяжелым течением вплоть до развития фульминантного гепатита (ФГ).
Нередко заболевание начинается незаметно с астеновегетативных проявлений, болей в области правого предреберья, незначительной желтухи. Однако у большинства больных АГ начало болезни острое, как при остром вирусном гепатите (ОВГ), и при осмотре пациента врач впервые выявляет признаки хронического гепатита (ХГ) — телеангиоэктазии, пальмарную эритему, увеличение печени и селезенки, а также изменения в анализах крови — гипергаммаглобулинемию, увеличение IgG, снижение содержания общего белка, резкое увеличение СОЭ. Лейкопения и тромбоцитопения наблюдаются у больных на поздних стадиях болезни или при развившихся гиперспленизме и синдроме портальной гипертензии.
Когда АГ впервые проявляется желтухой, как при ОВГ, приходится дифференцировать его от гепатитов А, В, Е и особенно С, при котором антитела в сыворотке крови могут появляться через достаточно продолжительное время после начала болезни. Желтуха у пациентов с АГ может быть разной степени выраженности, часто появляется на поздних стадиях заболевания, бывает непостоянной и усиливается в период обострений. В общем же у большинства больных чаще всего изменяются аминотрансферазы, нежели щелочная фосфотаза (ЩФ) или билирубин.
| Аутоиммунный гепатит был выделен из группы болезней печени и впервые описан как отдельная нозология в начале 50-х годов [33]. В научной литературе существовал под разными названиями. Термин люпоидный гепатит, который часто использовался в нашей стране, ввел в 1956 году Дж. Маккей с соавторами в журнале Lancet, так как при этом заболевании нередко в сыворотке крови больных выявлялись волчаночные клетки. Потом, в последующие годы, люпоидный, или классический, АГ стали называть аутоиммунным активным хроническим гепатитом, но в 1993 году Международная группа по изучению болезней печени предложила термин АГ, а также критерии установления его диагноза [17] |
Для АГ характерно поражение кожи в виде геморрагической сыпи, оставляющей после себя пигментацию. Из других симптомов встречаются волчаночная и узловатая эритемы, очаговая склеродермия, пальмарная эритема и телеангиоэктазии. У всех больных выявляются изменения в эндокринной системе — аменорея, угри, гирсутизм, стрии. Диагностическое значение отдельных симптомов болезни при АГ неодинаково. К наиболее значимым относятся длительная лихорадка и арталгии. В большинстве случаев АГ они присутствуют одновременно, являясь наиболее частыми и постоянно встречающимися жалобами больных [4].
Один из вариантов начала АГ — появление лихорадки с внепеченочными проявлениями, из которых следует назвать аутоиммунный тиреоидит, язвенный колит, гипертиреоидизм, гемолитическая анемия, идеопатическая тромбоцитопения, сахарный диабет, целиакия, полимиозит, фиброзирующий альвеолит, гломерулонефрит и т. д. Желтуха при этом варианте появляется позже [20].
Часто АГ сопровождается бесплодием, однако при возникновении беременности и последующих родах на фоне компенсированного процесса это не влияет на течение АГ и судьбу ребенка даже при постоянном приеме преднизолона (ПР) [30]. Беременность на стадии сформировавшегося ЦП и синдрома портальной гипертензии, которые выявляются у трети больных на момент выявления АГ, нежелательна [3].
В отличие от ХВГ течение АГ у больных непрерывно прогрессирующее, без самопроизвольных ремиссий. Улучшения самочувствия бывают кратковременными, нормализации биохимических процессов не происходит. Прогноз течения АГ хуже у пациентов с острым началом болезни по типу ОВГ, с наличием признаков холестаза, асцитом, повторными эпизодами острой печеночной энцефалопатии (ОПЭ). Как правило, больные, пережившие критический период, имеют лучший прогноз.
Диагноз АГ выставляется на основании соответствия лабораторных и гистологических данных, отсутствия маркеров ВГ, исключения злоупотребления алкоголем и контактов с препаратами крови, гипотоксическими веществами, повышения гамма-глобулинов не менее чем в 1,5 раза выше нормы. Повреждение желчных протоков, отложение меди, гемосидероз, при которых также могут выявляться ЛГ и ступенчатые некрозы, предполагают другую причину ХГ и исключают диагноз АГ. ANA, SMA и LRM-1 должны быть в титрах не менее 1:80 у взрослых и 1:20 у детей (рекомендации Международной группы, 1993).
Дифференциальный диагноз между АГ и другими аутоиммунными заболеваниями, в основном первичным билиарным циррозом (ПБЦ), первичным склерозирующим холангитом (ПСХ), ХВГ основывается на клинических, гистологических и иммунологических параметрах. Однако нередко выявляется так называемый overlap-синдром, когда одновременно у пациентов выявляются признаки АГ и вышеперечисленных хронических заболеваний печени. Далее они будут описываться как варианты АГ [7, 13]. Предполагаемый диагноз АГ в данном случае подразумевает сходство с клиникой АГ (жалобы на слабость, арталгии, миалгии), а биохимический анализ крови отражает преимущественно изменения холестатического порядка, имеет место кожный зуд разной степени выраженности. Пациенты с такими вариантами АГ могут быть обоего пола, любого возраста, но все же чаще это женщины в возрасте до 40 лет и моложе. На гистологии находят перипортальный гепатит с или без ЛГ, часто с поражением желчных протоков, жировой дистрофией гепатоцитов и лимфоидной инфильтацией портальных трактов в виде гранулем [7, 10].
Деление АГ на подтипы практического значения не имеет, однако следует иметь в виду, что подтип 2 АГ может быть связан с гепатитом С либо HCV может индуцировать появление АГ у генетически предрасположенных лиц. Нет данных о различиях гистологической картины при отдельных подтипах АГ
Большинство больных с ПБЦ можно точно отделить от пациентов с АГ с помощью характерных лабораторных и иммунологических данных. Однако при этом варианте наряду с характерными параметрами АГ нередко выявляются гистологические признаки холангита и АМА (антитела к антигенам внутренней поверхности мембраны митохондрий), что очень характерно для ПБЦ. Наиболее важным для подтверждения диагноза ПБЦ является обнаружение АМА подтипа М2 [6]. АМА выявляются у 20-27% больных АГ в разных титрах [19]. Это может отражать диагностические ошибки в определении иммуносерологических маркеров, другие заболевания или одну из стадий ПБЦ. Если у больного повышена щелочная фосфатаза (ЩФ), IgM сыворотки крови и обнаружена АМА — вероятен диагноз ПБЦ. Трех-шестимесячный курс лечения стероидами помогает расшифровать преобладающую патологию — при реакции на лечение можно говорить о превалировании АГ.
Установлено, что у 16% больных АГ выявляется язвенный колит (ЯК), наличие которого характерно для пациентов с ПСХ (от 40 до 60% больных). К тому же при таком сочетании — АГ и признаки ПСХ (наличие ЯК, поражение желчных протоков, слабый ответ на стероиды) — также обнаруживают фенотип HLA-B8, HLADR3, HLA DR4. Поэтому наличие кожного зуда у больных АГ и повышение ЩФ более чем в четыре раза против нормы указывают на необходимость проведения холангиографии (ХГР) и вероятность развития варианта АГ и ПСХ. Поражения желчных протоков несовместимы с диагнозом АГ. Они редки, но когда появляются у больных АГ с сопутствующей патологией кишечника или атипичным повышением ЩФ, можно допустить этот вариант АГ. Окончательный диагноз зависит от результатов ХГР. ХГР выявляет признаки склерозирующего холагнита у 42% больных АГ и ЯК. Но иногда ХГР бывает в норме у 14% больных ПСХ при гистологически подтвержденном диагнозе. Об этом необходимо помнить [24].
АГ считается заболеванием невирусной этиологии, но у 4% больных АГ выявляются антиHCV и еще у 4% — маркеры вируса гепатита В. Больные АГ, имеющие атипичное течение болезни либо плохо отвечающие на терапию стероидами, нередко имеют в сыворотке крови HCV RNA. Любопытно, что 11% больных ХВГ имеют SMA и 28% — ANA. У 62% выявляются аутоантитела к щитовидной железе и ревматоидный фактор. Большая часть этих больных имеют низкие титры SMA и ANA (1:80 и ниже), а пациенты с точным диагнозом АГ — SMA в титрах 1:160 и ANA 1:320. Поэтому больные АГ и с выявляемыми SMA или ANA в титрах ниже 1:320 могут быть отнесены к группе с превалированием вирусного заболевания [11].
Тем не менее пациенты с АГ имеют более выраженную инфильтрацию портальных трактов плазматическими клетками, более выраженные воспалительные изменения в дольках и больше ступенчатых и перисептальных некрозов по сравнению с пациентами ХВГ, особенно ХГС. У больных ХВГ/ХГС наоборот — в портальных трактах преобладает лимфоидноклеточная инфильтрация, чаще выявляется стеатоз и повреждения желчных протоков, особенно при ХГС.
У 13% взрослых больных с признаками АГ не обнаруживаются аутоантитела, а все остальные признаки — иммунологические, биохимические и гистологические, а также возраст и пол соответствуют критериям постановки диагноза АГ. Что важно, эти больные также хорошо реагируют на лечение стероидами [8, 9]. Отмечено, что с течением времени при динамическом наблюдении у некоторых из них появляются соответствующие аутоантитела, характерные для АГ.
Несмотря на разнообразие клинической картины, при АГ основой лечения является назначение преднизолона (ПР). Ответ на данную терапию — один из критериев постановки диагноза АГ. Целесообразность назначения ПР при АГ доказана в многочисленных исследованиях и обусловлена редкими самопроизвольными ремиссиями в течении болезни, высокой смертностью и ухудшением качества жизни [12, 18, 23, 28, 29]. При назначении ПР смертность удается снизить в течение пяти лет с 50 до 20%, а частоту индуцированных ремиссий довести до 80%. У большинства больных ремиссии появляются в течение первых двух лет терапии и почти у всех в последующие четыре года лечения.
Лечение ПР следует назначать всем больным АГ высокой степени активности с фиброзом и циррозом или без. У больных с умеренной степенью активности болезни назначение ПР часто определяется наличием жалоб и симптомов болезни. Больные без симптомов и с умеренной степенью активности процесса по гистологической картине не нуждаются в лечении, но должны тщательно и регулярно наблюдаться для своевременного выявления признаков прогрессирования болезни.
Как правило, начальная доза ПР составляет 20-30 мг/сутки с последующим постепенным снижением ее до поддерживающей — обычно 10 мг/сутки. Из всех схем лечения предпочтителен ежедневный прием однократно утром. Осложнения терапии наблюдаются при дозе более 10 мг/сутки. Нет точных рекомендаций по отмене или снижению дозы иммуносупрессоров, некоторые больные могут долго оставаться в ремиссии после отмены ПР.
Однако было установлено, что у большей части больных в дальнейшем, даже спустя несколько лет после ремиссии, появляются признаки обострения и часто требуется большая доза для ее достижения [15].
Комбинация ПР с азатиоприном (АЗА) может уменьшить побочные эффекты (при этом требуется небольшая доза ПР). Лучше давать 10 мг/сутки ПР с 50 мг/сутки АЗА, чем один ПР, но в большей дозе. Сам АЗА не способен индуцировать ремиссию, но его добавление к ПР поддерживает ее даже в дозе 1 мг/кг/сутки. При неэффективности лечения АЗА назначали 6-меркаптопурин с хорошим эффектом [25]. У 20% больных АГ не удается достигнуть ремиссии — чаще всего у пациентов с признаками ЦП, лиц молодого возраста, при длительном анамнезе болезни до начала терапии ПР и у больных с фенотипом HLA-B8, DR3 [28]. Побочные эффекты при назначении иммунодепрессантов редкие, это в основном диспепсический синдром, сыпи, кушингоидизм, нарушение роста и развития у детей, сахарный диабет и остеопороз у женщин в менопаузе. АЗА может индуцировать миелосупрессию, возникновение катаракты, обладает онкогенным и, возможно, тератогенным эффектами.
Лечение вариантов АГ представляет определенные трудности. Основа терапии, препарат выбора для начала лечения — и здесь ПР. При сочетании АГ и ПБЦ назначают ПР в дозе 20 мг/сутки от трех до шести месяцев, а при отсутствии эффекта — урсодезоксихолевую кислоту (УДХК) или ее коммерческие препараты (урсофальк, урсосан, урсодиол и др.) по 13-15 мг/сутки от трех до шести месяцев.
Тактика лечения больных с вариантом АГ и ПСХ та же, что и при АГ и ПБЦ. Больные АГ и ЯК отвечают на терапию ПР хуже, чем больные с одним АГ (не столь часты ремиссии, чаще и быстрее выявляют прогрессирование к ЦП). Эти пациенты, возможно, должны лечиться УДХК большими дозами (до 15-20 мг/кг/сутки), если признаки холестаза выражены.
При сочетании АГ и ХВГ назначают ПР 20 мг/сутки или 10 мг/сутки ПР и 50 мг/сутки АЗА на три–шесть месяцев, если превалируют признаки АГ. Рекомбинантный ИФН в дозе 3 млн. МЕ/сутки три раза в неделю до 6 месяцев назначают при выявлении признаков ХВГ и маркеров репликации вируса либо неэффективности стероидной терапии [21, 5]. Лечение таких больных представляет собой сложную задачу, так как ПР усиливает вирусную репликацию, а ИФН может усилить иммуноопосредованный печеночно-клеточный некроз, перевести ХВГ в АГ, который до этого мог быть в латентном состоянии, обострить течение болезни с развитием внепеченочных аутоиммунных проявлений, индуцировать выброс антител с неясным клиническим значением. Поэтому лечение состоит в правильном определении преобладания тех или иных клинических синдромов или признаков. В любом случае обострение болезни печени или внезапное появление признаков аутоиммунного заболевания у пациентов с признаками АГ, но с преобладанием вирусного поражения указывает на необходимость прерывания лечения ИФН.
Тактика лечения больных криптогенным ХГ состоит в назначении ПР 10-20 мг/сутки вместе с 50 мг/сутки АЗА до появления ремиссии или максимального эффекта.
Литература
Описаны методы лечения первичного склерозирующего холангита (ПСХ). Исследованы распространенность и спектр внепеченочных проявлений ПСХ, наиболее часты из них – хронические воспалительные заболевания кишечника. Установлено, что ПСХ у детей сопровождается
Mehtods of sclerosing cholangitis treatment have been described. Prevalence and the range of out-hepatic preparations of primary sclerosing cholangitis have been researched including the most frequent - bowels' chronic inflammatory diseases. Primary sclerosing cholangitis of children is more frequently followed by system presentations that of adults.
Первичный склерозирующий холангит (ПСХ) — это хроническое прогрессирующее холестатическое заболевание печени неясной этиологии, характеризующееся негнойным деструктивным воспалением, облитерирующим склерозом и сегментарной дилятацией внутри- и внепеченочных желчных протоков, приводящее к развитию вторичного билиарного цирроза, портальной гипертензии и печеночной недостаточности [1]. Распространенность ПСХ в разных странах отличается. Так, в Северной Европе она составляет 10 на 100 тысяч населения, а в Азии ПСХ встречается в 10–100 раз реже. Болезнь развивается чаще у мужчин (женщины болеют в 1,5–2 раза реже) [2]. Чаще всего заболевание начинается в возрасте 30–40 лет, хотя известны случаи развития ПСХ в детском и пожилом возрасте [3].
В патогенезе ПСХ важная роль принадлежит нарушению иммунной толерантности. Доказательством этого являются обнаружение широкого спектра аутоантител в сыворотке крови таких больных: обнаружены антитела к ДНК, к гладкой мускулатуре, к билиарному эпителию, к тиреопероксидазе, антимитохондриальные антитела и некоторые другие (описано более 25 видов аутоантител у больных ПСХ), а также частое сочетание ПСХ с другими аутоиммунными заболеваниями, которые могут быть рассмотрены как системные проявления ПСХ [4]. Хронические воспалительные заболевания кишечника обнаруживают у 80–90% больных ПСХ, чаще это язвенный колит, реже встречается болезнь Крона с поражением толстой кишки [2, 3]. Реже ПСХ сочетается с аутоиммунным гепатитом, аутоиммунным тиреоидитом, сахарным диабетом 1-го типа, псориазом (до 10% случаев). Описаны отдельные наблюдения развития ревматоидного артрита, системной склеродермии, системной красной волчанки, склерита, увеита у больных ПСХ [5]. Частое сочетание ПСХ и воспалительных заболеваний кишечника легло в основу предположения о триггерной роли бактериальных антигенов в возникновении ПСХ, однако доказать это не удалось [2]. У родственников больных ПСХ в 11,5 раз повышен риск развития ПСХ, в 3 раза — язвенного колита и в 1,4 раза — болезни Крона [6]. Было сделано предположение о ключевой роли некоторых генов в развитии ПСХ. Были проведены исследования генов HLA-A, HLA-B, HLA-DR, MST1R, NOD2, ATG16L1, однако какого-либо одного гена, специфичного для ПСХ, не было найдено [2].
Препаратом выбора при консервативном лечении ПСХ является урсодезоксихолевая кислота в дозе 20–30 мг/кг/сут (Урсофальк, Урсосан, Урдокса). Урсодезоксихолевая кислота обладает гепатопротективным действием — она образует нетоксичные мицеллы с токсичными желчными кислотами (хенозедоксихолевой, литохолевой), а также включается в состав мембран гепатоцитов и холангиоцитов, увеличивая их стабильность. Это уменьшает степень повреждения клеток печени токсичными желчными кислотами. Урсодезоксихолевая кислота обладает свойствами иммуномодулятора, угнетая экспрессию HLA-антигенов на мембранах гепатоцитов и холангиоцитов. Кроме того, препарат уменьшает литогенность желчи за счет уменьшения абсорбции холестерина в кишечнике, а также стимулирует холерез, что увеличивает выведение желчных кислот через кишечник [2, 3]. При неэффективности монотерапии урсодезоксихолевой кислотой к терапии добавляют фибраты, которые способствуют уменьшению холестаза [2].
Получены данные о том, что лактулоза (Дюфалак, Нормазе) в дозе 15–45 мл/сут обладает гепатопротективным действием, которое может реализовываться посредством нескольких механизмов: 1) ингибирование продукции аммиака и других ксенобиотиков; 2) утилизация образовавшегося аммиака; 3) нарушение всасывания и быстрое выведение аммиака с калом [7, 8]. Уменьшение образования аммиака в кишечнике под действием лактулозы связано с уменьшением количества кишечной микрофлоры, продуцирующей уреазу. Утилизация аммиака происходит при помощи бактерий, использующих азотсодержащие соединения в качестве субстрата для синтеза собственных белков; лактулоза способствует увеличению количества этих микроорганизмов в кишечнике. Кроме того, лактулоза способна связывать молекулы аммиака и увеличивать скорость транзита содержимого по кишечнику, что способствует быстрому выведению аммиака из организма. Лактулоза обладает свойствами пребиотика: под ее воздействием в кишечнике увеличивается количество бифидобактерий и уменьшается количество патогенных бактерий, что предотвращает избыточный бактериальный рост и снижает риск проникновения антигенов патогенных бактерий из кишечника в кровь [14]. Недавно в России было проведено исследование, согласно результатам которого при лечении больных с холестазом урсодезоксихолевой кислотой в комбинации с лактулозой (Урсолив) наблюдается большее уменьшение выраженности холестаза, чем при лечении только урсодезоксихолевой кислотой [9].
Таким образом, совместное применение урсодезоксихолевой кислоты и лактулозы приводит к синергизму гепатопротективного эффекта обоих веществ.
При сочетании ПСХ с аутоиммунным гепатитом к лечению добавляют глюкокортикостероиды и/или иммуносупрессанты в дозах, стандартных для лечения аутоиммунного гепатита [9]. С целью устранения зуда в качестве препарата первой линии применяют Холестирамин (4 г/сут), однако не все пациенты хорошо его переносят: наиболее частыми причинами отказа от препарата являются его вкус, вздутие живота, запоры или диарея. При непереносимости Холестирамина применяют Рифампицин (300 мг/сут), Сертралин (75–100 мг/сут), Налтрексон (50 мг/сут). При выраженном зуде может потребоваться альбуминовый диализ или плазмаферез. Нестерпимый зуд, не купирующийся консервативными методами, может являться самостоятельным показанием к трансплантации печени. Длительный холестаз является фактором риска развития остеопороза, поэтому таким пациентам в схему лечения необходимо включать кальций (1500 мг/сут) и витамин D (1000 МЕ/сут). Витамин K назначается при наличии геморрагического синдрома, особенно перед инвазивными диагностическими процедурами [3, 10].
К хирургическим методам лечения ПСХ относятся установка стентов, резекция пораженных участков желчных протоков, трансплантация печени. Стентирование, баллонная дилятация и резекция пораженных участков протоков лишь временно уменьшают выраженность холестаза, не останавливая прогрессирование ПСХ. Наиболее эффективным хирургическим методом лечения ПСХ является трансплантация печени, которая улучшает выживаемость больных. Однако у 15–20% больных развивается рецидив ПСХ в трансплантате, что ухудшает прогноз таких пациентов [2, 3, 10].
При естественном течении ПСХ часто развивается холангиокарцинома (до 40% больных), повышен риск развития рака поджелудочной железы и колоректального рака, поэтому необходимы как можно более ранние диагностика и лечение этого заболевания [11, 12].
Цель исследования. Изучение распространенности и спектра внепеченочных проявлений первичного склерозирующего холангита.
Материалы и методы. Объектом исследования были 23 больных первичным склерозирующим холангитом, наблюдавшихся в Клинике нефрологии, внутренних и профессиональных заболеваний им. Е. М. Тареева Первого МГМУ им. И. М. Сеченова в период с 1984 по 2010 гг. В группе было 12 мужчин и 11 женщин, средний возраст данной группы составил 36,4 ± 13,7 года, средний возраст начала заболевания 30,6 ± 15 лет, средняя продолжительность заболевания 5,8 ± 5,2 года.
Материалами исследования служили данные из архивных записей в историях болезни и амбулаторных картах. Критерием включения в исследование являлся подтвержденный диагноз ПСХ (морфологически, посредством магнитно-резонансной холангиографии или ретроградной эндоскопической холангиопанкреатографии). Критерием исключения являлось наличие у больного сочетания ПСХ и аутоиммунного гепатита, так как аутоиммунный гепатит также характеризуется широким спектром системных проявлений. Изучалось наличие следующих системных проявлений ПСХ: хронические воспалительные заболевания кишечника (язвенный колит и болезнь Крона), поражение глаз (склерит, кератит), аутоиммунный тиреоидит, синдром Шегрена, геморрагический васкулит.
Результаты и обсуждение. У 16 больных (69,6%) были выявлены внепеченочные проявления ПСХ. Из данных, представленных в табл. 1, видно, что в обследованной группе были выявлены хронические воспалительные заболевания кишечника (язвенный колит и болезнь Крона), синдром Шегрена, геморрагический васкулит, поражение глаз (склерит, кератит), поражение щитовидной железы (аутоиммунный тиреоидит).
У пациентов с сочетанием ПСХ и болезни Крона (5 больных) в 1 случае была поражена только толстая кишка; в 2 случаях — и тонкая, и толстая кишка; в 2 случаях — только тонкая кишка. Отсутствие поражения толстой кишки у двух больных с болезнью Крона при ПСХ указывает на необходимость длительного динамического наблюдения за этими пациентами, так как в дальнейшем у них возможно присоединение поражения толстой кишки.
У 4 больных (17,4%) отмечено сочетание нескольких системных проявлений ПСХ. У 3 больных выявлено два внепеченочных проявления ПСХ (2 случая — язвенный колит в сочетании с аутоиммунным тиреоидитом, 1 случай — язвенный колит в сочетании с геморрагическим васкулитом). У 1 пациента выявлено три внепеченочных проявления ПСХ — язвенный колит в сочетании с геморрагическим васкулитом и поражением глаз (склерит, кератит).
В связи с повышенным риском развития онкологических заболеваний у больных ПСХ мы изучили распространенность злокачественных заболеваний в изучаемой группе больных. Онкологические заболевания были выявлены у 3 больных (13%): 1 случай (пациент с ПСХ в сочетании с болезнью Крона) — лимфома с поражением тонкой кишки, выявлена одновременно с подтверждением диагноза ПСХ; 1 случай (больная с ПСХ без системных проявлений) — лимфома с поражением печени, лимфатических узлов ворот печени, забрюшинных лимфатических узлов, выявлена одновременно с подтверждением диагноза ПСХ; 1 случай (пациентка с ПСХ в сочетании с язвенным колитом) — аденокарцинома толстой кишки, выявлена через 13 лет после подтверждения диагноза ПСХ. В связи с частым развитием злокачественных опухолей у больных ПСХ необходима постоянная онкологическая настороженность врачей, работающих с этой категорией больных.
Также мы проанализировали распространенность системных проявлений ПСХ в зависимости от пола больных и возраста первых проявлений ПСХ (табл. 2 и 3).
С целью изучения связи частоты обнаружения системных проявлений ПСХ и возраста начала заболевания мы разделили больных ПСХ на две подгруппы — тех, у кого заболевание началось в детском возрасте (6 человек, возраст начала заболевания от 10 до 17 лет), и тех, у кого первые проявления ПСХ возникли во взрослом состоянии (17 человек, возраст начала заболевания от 20 до 66 лет) (табл. 3).
Из табл. 3 следует, что в группе больных, у которых ПСХ начался в детском возрасте, внепеченочные проявления регистрировались чаще, чем в группе пациентов, заболевших во взрослом состоянии. Причина такого различия распространенности системных проявлений ПСХ в различных возрастных группах в настоящее время неясна, необходимы дальнейшие исследования этого вопроса, в том числе обследование больших групп больных ПСХ.
Выводы
- ПСХ часто сопровождается внепеченочными проявлениями, наиболее распространенные из которых — хронические воспалительные заболевания кишечника.
- У одного больного может встречаться сочетание нескольких системных проявлений ПСХ.
- Поражение кишечника при ПСХ чаще развивается у мужчин, поражение щитовидной железы — у женщин.
- ПСХ, развившийся в детском возрасте, сопровождается системными проявлениями чаще, чем ПСХ, начавшийся у взрослых.
Литература
Е. А. Александрова*
Э. З. Бурневич*, кандидат медицинских наук, доцент
Е. А. Арион**
* ГБОУ ВПО Первый МГМУ им. И. М. Сеченова Минздравсоцразвития РФ,
**Клиника нефрологии, внутренних и профессиональных болезней им. Е. М. Тареева, Москва
Читайте также: