
Как сделать макет вируса своими руками
Обновлено: 15.04.2024
Можем поиграться с библиотекой для начала, начнём с инициализации флэшки как HID-устройства методом begin().

Отлично. Создадим эксплоит во фреймворке Metasploit.
Будем использовать модуль web_delivery. Я выбрал его из-за высокой скорости и низкой вероятности срабатывания антивируса. Он также ничего не пишет на диск, так что не оставит следов по окончании работы.
Здесь мы ломаем 64-битную Windows 10, так что выберем мишенью PowerShell, но имейте в виду, это не эксплоит против PowerShell. Мы просто используем оболочку, чтобы скачать нужные файлы с сервера.
Нужно сказать нашей программе, откуда брать бинарники:
Дальше указываем порт, который не вызовет подозрений, что насчёт 443? ;)
Metasploit каждый раз генерирует случайный URIPATH, а мы хотим иметь возможность запускать и останавливать прослушку порта в любой момент без необходимости перекомпилировать код для флешки.
Теперь нужно выбрать Powershell в качестве метода доставки. Эксплоит поддерживает три цели, помеченные идентификаторами: это 0: Python, 1: PHP, и 2: Powershell.
И наконец exploit .
Чтобы удобно было останавливать и возобновлять прослушку порта, создадим конфигурационный файл: usb.rc.
Получаем полезную нагрузку для запуска на компьютере жертвы:
Теперь можем запустить это с флэшки.
Работает очень неплохо. Нам нужно около 40 секунд, чтобы поиметь Дейнерис, я имею в виду компьютер жертвы.

Я выбрал одну из тех неприметных USB-флешек, которые рекрутеры раздают миллионами, и заказал эти классные маленькие OTG-адаптеры microUSB − USB A. Пришлось отрезать ненужные части печатной платы, чтобы она поместилась в корпус, всунул OTG-адаптер в корпус USB A и заклеил всё суперклеем. По мне так выглядит вообще не подозрительно, но всё-таки 10 секунд — это немалое время, особенно когда прячешься от драконов.

Вы также можете заказать Arduino Pro Micro на Amazon примерно за $10. Если есть терпение, то можно даже найти на eBay примерно за $3 или $4. У меня не нашлось USB-флэшки достаточно большого размера для Pro Micro, так что я подключил OTG-адаптер, перемотал его изолентой и на этом успокоился.

Нужно немного изменить программу, потому что мы используем другую библиотеку, но работать она будет как и раньше.
Самое большое преимущество Pro Micro — это скорость. Теперь нам нужно всего 3 секунды физического доступа. Настоящая атака на ходу. Если вы намерены применить эту силу, делайте это ради благого дела. Убейте Серсею.
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.

Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.

Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Белки: как получают, хранят и отображают информацию об их структуре?
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.

Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.
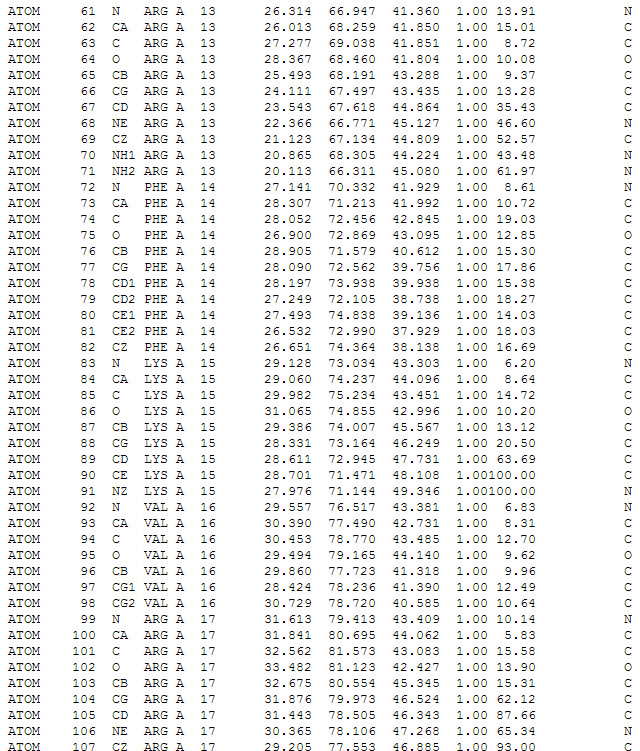
Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.

Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:
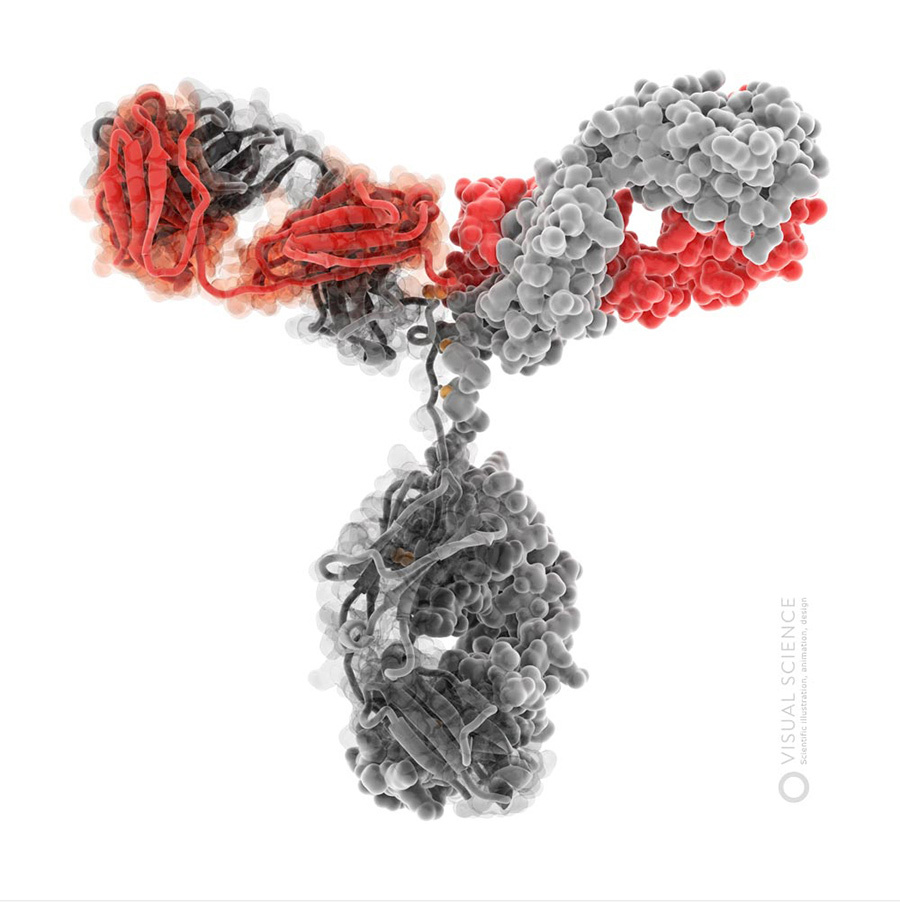

Изображения молекулы иммуноглобулина G.
Молекулярное моделирование

Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Докинг
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Посттрансляционные модификации
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Молекулярная динамика и оптимизация структур
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.
Молекулярный биолог Валерия Архипова и CGI художник Алексей Солодовников создали атомарную модель вируса SARS-CoV-2 и анимировали ее. По просьбе N + 1 они рассказывают, как много сил и времени заняла у них эта работа и какие данные им потребовались, чтобы построить изображение, которое одновременно было бы достоверным и при этом красивым.

Атомарная модель (каждый шарик — атом) SARS-CoV-2. Цветами обозначены: S- (бирюзовые), M- (зеленые) и E-белки (красные); мембрана (синий) и олигосахариды (оранжевые)
Наш вирус, помимо молекул оболочки (о том, почему они именно такие, подробнее расскажем ниже), собран из белков трех типов и олигосахаридных остатков на их поверхности.
S-белки (бирюзовые), они же спайк-белки или белки шипа — их вирус использует для того, чтобы прикрепляться к клеткам. Если эти белки свою работу делают хорошо, то вирус пробирается в клетку и переходит к размножению внутри нее.
Е-белки (красные), взаимодействуют с другими структурными белками, а также, будучи трансмембранными белками, образуют ионный канал, тем самым помогая сборке новых вирионов в комплексе Гольджи клетки-хозяина.
Другие модели
Изображения вируса, которые можно найти в открытом доступе, делятся на два типа: художественные иллюстрации без опоры на молекулярную биологию и работы ученых, где в приоритете — научная достоверность, а не эстетическая привлекательность.
Мы подумали, что можем совместить эти подходы: соблюсти достоверность и одновременно сделать изображение насыщенным. Показать и сложность, и красоту.
Конечно, работали мы не в информационном вакууме. Есть и другие модели — они нас вдохновляли и учили. Нам хотелось сделать не хуже или добавить что-то свое. Вот на какие модели мы обратили внимание, прежде чем заняться своей:
Это видео показало, что мы можем сделать нашу модель цветной, эстетически привлекательной. Выделить цветом разные части вируса, что улучшает восприятие изображения. Плюс здесь мы впервые увидели движение вируса — на изображениях, которые мы видели до этого, вирус всегда выглядел статично.
Данная работа показала, что мы можем уточнить структуру самого вируса, сделать ее атомарной. Это многократно повысило сложность модели, но и ее достоверность также возросла.
Тут мы увидели, что количество ключевых белков у вируса может быть очень различным. Мы приняли решение сосредоточиться на уточнении их количества, отдавая предпочтение самым новым источникам.
Эта работа начинается с 2D-иллюстрации ученого и художника Дэвида Гудселла (David S. Goodsell). Мы увидели, насколько важно придумать и выдержать цветовое решение модели вируса. В итоге, прежде чем мы дошли до финала, было отвергнуто 11 разных цветовых решений.
И последняя работа — это модель студии Visual Science. Эта модель показала, что наше решение сделать атомарную модель вируса — совершенно правильное. Также мы поняли, что число ключевых белков на поверхности вируса может сильно варьироваться. Мы приняли решение сосредоточиться на уточнении их количества, отдавая предпочтение самым новым научным источникам. Также мы увидели новую крупную задачу — сделать модель внутренней части вируса. Эту задачу мы оцениваем, как более сложную и не менее важную.
Все модели выше в большинстве своем сделаны крупными научными коллективами с привлечением значительных вычислительных ресурсов (облака, фермы, кластера). Нам было интересно, что может сделать наша команда из двух человек.
Источники данных
Часть белков на нашей иллюстрации взяты из базы Protein DataBase. Каждый белок — это отдельный файл, внутри которого хранятся координаты атомов белка, а также дополнительная информация: химический состав, аминокислотный, интенсивность колебаний атомов и тому подобное.
Для разных белков в Protein Database используют разные подходы к помещению атомов исследуемой молекулы в систему координат. Так, например, в прямоугольных декартовых координатах ось Z совмещается с максимальным значением момента инерции (для горизонтального расположения фрагмента белка) или совпадает с минимальным значением момента инерции (в случае вертикального расположения). Другой вариант используют для исследования длинных спиралевидных молекул: при этом ось X соответствует среднему значению момента инерции эллипсоида. Также широко используются сферические, цилиндрические и другие системы координат, которые потом пересчитываются в декартовы при создании PDB-файла.
По сути, формат PDB — это текстовый файл с данными про каждый атом. Вот как записывают, например, структуру Е-белка 5X29:

Фрагмент pdb-файла белка Е-белка 5X29. Например, для 878 атома координаты 0.035 -9.540 8.679 — это кислород
А вот как эта же информация выглядит уже в Houdini — программе, в которой данные из pdb-файлов преобразуются в 3D-модель, а затем в иллюстрацию.

Е-белок 5X29 в Houdini. Каждая точка — атом
PDB — очень ценный источник информации, без него у нас бы ничего не получилось. Это огромная — 178 938 биологических макромолекулярных структур на июнь 2021 года — международная база, которую пополняют ученые со всего мира, выкладывая данные своих исследований в открытый доступ.
Молекул оболочки нового коронавируса в полном виде в библиотеке PBD нет. Мы взяли оттуда липиды, из которых она может состоять, и построили ее самостоятельно. Вирус собирает свою оболочку из фрагментов мембраны клетки-хозяина — этот суперкапсид способствует повышению заразности вируса. Суперкапсид нашего вируса построен из липидов мембраны клетки эпителия.
Далее нам нужно было определить количество объектов, из которых состоит вирус, и предсказать их движение. В нашей модели мы отобразили все атомы, которые есть в исходных данных, без упрощения. Вот что у нас получилось сначала:

Промежуточная модель коронавируса SARS-COV-2
Затем нам удалось получить консультацию экспертов. Это позволило повысить точность нашей модели. Мы чрезвычайно признательны Николаю Никитину с кафедры вирусологии МГУ и Софье Борисевич из лаборатории химической физики Уфимского Института химии РАН. Вместе с ними мы уточнили количество S-белков, которое зависит от размера вириона (сам он составляет 50-200 нанометров). По последним данным их не 90, как было в первой версии нашей иллюстрации, а меньше — от 26 до 41-го. На нашей модели S-белков 38.
Также большим сюрпризом для нас оказалась подвижность S-белка:

Иллюстрация, показывающая сгибы и вращение S-белка
Choi et al. / Journal of Chemical Theory and Computation, 2021
Помимо этого, мы выяснили, что M-белки расположены на оболочке намного плотнее — их стало около 1000 штук на вирион. А E-белков, как минорной компоненты, напротив, очень мало. В нашем случае их 15 штук на вирион.
Как моделировали
Вот как выглядит 3D-модель вируса в Houdini:

Houdini. Собранная 3D модель. Геометрическое представление. Без материалов и света
Houdini используется для создания спецэффектов в кино и рекламе: взрывы, дым, вода, огонь, разрушения. Подошел он и для наших задач. Внешняя часть вируса нашей модели — приблизительно 25 000 000 атомов. Просчет изображения с материалами и светом выполнялся на GPU рендере RedShift. По сравнению с CPU (16-ядерный AMD против 3080 NVIDIA), прирост по скорости около семи раз. Модель вируса может быть посчитана в очень больших разрешениях. Рендер для фрагмента, на котором хорошо видны атомы, сделан в 8000х8000 пикселей, что эквивалентно 1300х1300 миллиметрам при печати в 150 точек на дюйм.
Вирус рендерился в черно-белом виде. И дальше уже по маскам красился вручную в программе для композитинга и монтажа — DaVinci Resolve. Такой подход позволил подобрать цветовую гамму без пересчета модели. Всего было испробовано порядка десяти вариантов цветовых решений. Ниже — не раскрашенная модель вируса.


Наша модель SARS-CoV-2. Фрагмент из полного изображения 8000х8000 пикселей (130х130 сантиметров). Хорошо видны атомы.
Итого
Моделированием внутренней структуры вируса мы не занимались. Это около 30 000 нуклеотидов РНК, N-белков и других элементов. Эта работа сложнее, чем моделирование внешнего вида вируса. Во многом это связано с отсутствием моделей белков и тем, что для этого нужно решить нетривиальную задачу укладки нити РНК в полости сферы вируса.
Повысить точность нашей модели можно с привлечением молекулярной динамики. Тогда оболочку можно будет изобразить точнее, как и само движение атомов. Молекулярная динамика позволяет получить очень точное представление о движении и взаимодействии частей вируса как друг с другом, так и с внешней средой. Но МД требует специальных навыков работы и очень требовательна к вычислительным ресурсам. У нас нет необходимых мощностей, чтобы работа была выполнена за приемлемые сроки. Поэтому движения атомов и частей вируса показаны условно.
В открытых источниках мы не нашли информации о структуре М-белка. Поэтому и он тоже показан условно. Размеры атомов усреднены.
Вся работа заняла у нас приблизительно полгода: она велась параллельно с другими проектами, после основной работы, домашних дел, с перерывами на болезни и праздники и поправкой на разные часовые пояса.
Мы получили модель вируса, изображения с ним и анимацию его условного движения. Что дальше, что с этим делать и какая от этого польза?
Во-первых, это красиво. Во-вторых, мы показываем сложность объектов на микроскопическом, атомарном уровне. Наша работа позволяет эту сложность увидеть. И последнее — пожалуй, самое важное. Наш проект показывает, что и один человек может сделать очень многое. А значит, может каждый из нас. Дерзайте!
Список литературы
1. Surya, W., Li, Y., Torres, J. Structural model of the SARS coronavirus E channel in LMPG micelles. // Biochim. Biophys. Acta - Biomembr. – 2018. – Vol. 1860. – N. 6. – P. 1309–1317.
2. Koppisetti, R. K., Fulcher, Y. G., Jurkevich, A., Prior, S. H., Xu, J., Lenoir, M., Overduin, M., Van Doren, S. R. Ambidextrous binding of cell and membrane bilayers by soluble matrix metalloproteinase-12. // Nat. Commun. – 2014. – Vol. 5. – P. 1–14.
3. Hillen, H. S., Kokic, G., Farnung, L., Dienemann, C., Tegunov, D., Cramer, P. Structure of replicating SARS-CoV-2 polymerase. // Nature. – 2020. – Vol. 584. – N. 7819. – P. 154–156.
4. Harris, L. J., Larson, S. B., Hasel, K. W., McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. // Biochemistry. – 1997. – Vol. 36. – N. 7. – P. 1581–1597.
5. Noreng, S., Bharadwaj, A., Posert, R., Yoshioka, C., Baconguis, I. Structure of the human epithelial sodium channel by cryo-electron microscopy. // Elife. – 2018. – Vol. 7. – P. 1–23.
6. Almond, A., DeAngelis, P. L., Blundell, C. D. Hyaluronan: The Local Solution Conformation Determined by NMR and Computer Modeling is Close to a Contracted Left-handed 4-Fold Helix. // J. Mol. Biol. – 2006. – Vol. 358. – N. 5. – P. 1256–1269.
7. Hurdiss, D. L., Drulyte, I., Lang, Y., Shamorkina, T. M., Pronker, M. F., van Kuppeveld, F. J. M., Snijder, J., de Groot, R. J. Cryo-EM structure of coronavirus-HKU1 haemagglutinin esterase reveals architectural changes arising from prolonged circulation in humans. // Nat. Commun. – 2020. – Vol. 11. – N. 1. – P. 1–10.
8. Yan, Renhong, Yuanyuan Zhang, Yaning Li, Lu Xia, Yingying Guo, Q. Z. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. // Science (80-. ). – 2020. – Vol. 3. – N. 3. – P. 1–8.
9. Javitt, G., Khmelnitsky, L., Albert, L., Bigman, L. S., Elad, N., Morgenstern, D., Ilani, T., Levy, Y., Diskin, R., Fass, D. Assembly Mechanism of Mucin and von Willebrand Factor Polymers. // Cell. – 2020. – Vol. 183. – N. 3. – P. 717-729.e16.
10. Daniel Wrapp, Nianshuang Wang, Kizzmekia S. Corbett, Jory A. Goldsmith, Ching-Lin Hsieh, Olubukola Abiona, B. S. G., McLellan, and J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. // Science (80-. ). – 2020. – Vol. 21. – N. 1. – P. 1–9.
11. Wang, M. Y., Zhao, R., Gao, L. J., Gao, X. F., Wang, D. P., Cao, J. M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. // Front. Cell. Infect. Microbiol. – 2020. – Vol. 10. – N. November. – P. 1–17.
12. Yao, H., Song, Y., Chen, Y., Wu, N., Xu, J., Sun, C., Zhang, J., Weng, T., Zhang, Z., Wu, Z., Cheng, L., Shi, D., Lu, X., Lei, J., Crispin, M., Shi, Y., Li, L., Li, S. Molecular Architecture of the SARS-CoV-2 Virus. // Cell. – 2020. – Vol. 183. – N. 3. – P. 730-738.e13.
13. Oostra, M., de Haan, C. A. M., de Groot, R. J., Rottier, P. J. M. Glycosylation of the Severe Acute Respiratory Syndrome Coronavirus Triple-Spanning Membrane Proteins 3a and M. // J. Virol. – 2006. – Vol. 80. – N. 5. – P. 2326–2336.
14. B.W. Neuman, M. J. B. Supramolecular Architecture of the Coronavirus Particle. // Adv. Virus Res. – 2020. – Vol. 96. – P. 1–27.
15. Neuman, B. W., Kiss, G., Kunding, A. H., Bhella, D., Baksh, M. F., Connelly, S., Droese, B., Klaus, J. P., Makino, S., Sawicki, S. G., Siddell, S. G., Stamou, D. G., Wilson, I. A., Kuhn, P., Buchmeier, M. J. A structural analysis of M protein in coronavirus assembly and morphology. // J. Struct. Biol. – 2011. – Vol. 174. – N. 1. – P. 11–22.
16. Yu, A., Pak, A. J., He, P., Monje-Galvan, V., Casalino, L., Gaieb, Z., Dommer, A. C., Amaro, R. E., Voth, G. A. A multiscale coarse-grained model of the SARS-CoV-2 virion. // Biophys. J. – 2021. – Vol. 120. – N. 6. – P. 1097–1104.
17. Yao, H., Song, Y., Chen, Y., Wu, N., Xu, J., Sun, C., Zhang, J., Weng, T., Zhang, Z., Wu, Z., Cheng, L., Shi, D., Lu, X., Lei, J., Crispin, M., Shi, Y., Li, L., Li, S. Molecular architecture of the SARS-CoV-2 virus. // Cell. – 2020. – Vol. 183. – N. 3. – P. 730–738.
18. Choi, Y. K., Cao, Y., Frank, M., Woo, H., Park, S. J., Yeom, M. S., Croll, T. I., Seok, C., Im, W. Structure, Dynamics, Receptor Binding, and Antibody Binding of the Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein in a Viral Membrane. // J. Chem. Theory Comput. – 2021. – Vol. 17. – N. 4. – P. 2479–2487.
Открытие вирусов
Вирусы были открыты в 1892 году Д.И. Ивановским случайно.

Он, изучая мозаичную болезнь табака, обнаружил в листьях больных растений инфекционное начало, которое с легкостью проходило через фильтр, задерживающий бактериальные клетки. Листья здоровых растений заболевают мозаичной болезнью, если в них внести фильтрат.
Аналогичные выводы в 1898 году сделал голландский микробиолог М. Бейеринк. Он высказал предположение, что мозаичную болезнь табака вызывают фильтрующие вирусы.
Размеры вирусов составляют от 15-2000 нм у растений, а у животных - 450 нм. Их микроструктуру можно увидеть только в электронный микроскоп, который изобрели в 30-е годы 20 века.
И что самое важно: мы знаем, как противостоять бактериям с помощью антибиотиков. А вот на вирусы они не действуют. Поэтому в 2014 году в Западной Африке произошла крупнейшая вспышка вируса лихорадки Эбола.
Вирус поражает человека, приматов и парнокопытных. Передача вируса происходит через прямой контакт с кровью или биологическими жидкостями, а также через микротравмы кожи. Заболевание начинается остро, с сильной слабости, острой головной боли, болей в мышцах, диареи, болей в животе. Позднее проявляется сухой кашель и колющие боли в грудной клетке, развивается обезвоживание организма, рвота. Появляется геморрагическая сыпь. В 40-50% случаев начинаются кровотечения из желудочно-кишечного тракта, носа, десен. В июле 2015 года ВОЗ сообщило об успешных тестах эффективной вакцины VSV-EBOV против лихорадки Эбола.
В 1917 году Феликс Д'эрелль выделил бактериофаги-вирусы бактерий. Но о них чуть позже.
Свойства вирусов
На основе многолетних исследований были определены такие свойства вирусов:
• неклеточное строение;
• облигатный паразитизм;
• отсутствие собственного роста и обмена веществ;
• отсутствие проявлений жизнедеятельности вне клетки-хозяина;
• использование органелл клетки-хозяина для синтеза новых вирусных частиц.
Строение вирусов
Вирусы - неклеточные формы жизни (организм не состоит из клеток). Вирусы имеют сравнительно простое строение.

Зика: что мы знаем об этом вирусе? Как уберечься от вируса? Вирус Зика передается человеку через укусы комаров рода Aedes, которые обитают в экваториальных и тропических лесах. После заражения резко поднимается температура, возникает тошнота и мигрень, развивается конъюктивит и появляется болезненная сыпь. ВОЗ официально объявила эпидемию лихорадки Зика. Только в 2016 году в Бразилии лихорадкой переболели 1,5 млн человек и количество зараженных продолжает расти. Вакцины против лихорадки не существует.
ДНК-содержащие вирусы
ДНК-содержащие вирусы вызывают папиллому человека, герпес, гепатит В.
ДНК-содержащие вирусы поражают также растения (золотая мозаика бобов, полосатость у кукурузы).
Бактериофаги
Среди вирусов есть наши друзья. Их называют бактериофагами. Это сложные вирусы, которые разрушают бактериальные клетки.
3. Вернитесь на сайт PDB и введите идентификационный код молекулы (PDB ID), например 2CAS в поле текстового ввода Enter (рис 3).
4. Изучите, каким образом осуществляется моделирование (рис 4,5). Полученный результат представьте в виде скриншота.
Компьютерная группа № 2.
Моделирование вирусов с помощью модульного оригами
Кусудама — объемное сферическое тело, сконструированное из бумажных цветов.

Основой кусудамы является правильный многогранник (куб, додекаэдр или икосаэдр).
Кусудамы – шары модульного оригами, которые могут наглядно иллюстрировать строения капсидов вирусов посредством модулей-заготовок.
Найдите в сети Интернет схемы таких кусудам и соберите фрагмент капсида вируса с помощью модулей.


Презентация на тему: Таинственный микромир или 3D - моделирование вирусов посредством online – программы Virus Particle Explorer и модульного оригами
Читайте также: