
Лимфома или вирусный лейкоз
Обновлено: 17.04.2024
Неврологические проявления лейкозов, лимфом у ребенка
Неврологические проявления часто возникают как начальные признаки или как поздние осложнения при лейкозах и лимфомах, и могут быть первым признаком (Aysun et al., 1994). На различных стадиях этих заболеваний часто наблюдаются сложные неврологические синдромы, приводящие к значительным трудностям в определении причины неврологических проявлений, так как они могут возникать вследствие самого заболевания, токсических осложнений лечения, вирусных или оппортунистических инфекций в результате иммуносупрессии, или вследствие геморрагических осложнений.
Тем не менее, от правильной диагностики неврологического нарушения зависит выбор соответствующего лечения.
а) Неврологические проявления лейкозов. Осложнения при лейкозах могут возникать вследствие лейкемической инфильтрации менингеальных оболочек, мозга или черепных нервов, либо вследствие кровоизлияний или инфекций.
Менингеальный лейкоз может возникать на любой стадии заболевания. От одной трети до половины детей пребывают в состоянии полной ремиссии, когда появляются неврологические осложнения. Лейкемическая инфильтрация первично поражает паутинную оболочку, а инфильтрация ткани мозга обнаруживается менее чем у 15% детей, погибших при рецидиве (Price, 1983). Особенности проявления лейкоза ЦНС включают головную боль, рвоту и отек диска зрительного нерва.
Судороги возникают примерно у 10% пациентов; риск припадков значительно выше у пациентов, получающих лечение метотрексатом. Повышение аппетита и набор веса могут возникать в результате инфильтрации гипоталамуса. Часто имеются параличи черепных нервов. КТ обычно не выявляет лейкемических инфильтратов (Chen et al., 1996), однако MPT с гадолинием может показать менингеальное вовлечение.
Крупные внутричерепные или параспинальные образования (гранулоцитарные саркомы или хлоромы) являются редкими (Brown et al., 1989). Они возникают при миелобластном лейкозе, что часто приводит к поражению позвоночного канала и компрессии спинного мозга и корешков. Поражения сетчатки, включая кровоизлияния и белые экссудаты, иногда с отеком диска, возникают примерно в 20% случаев лейкозов (Taylor, 1990).
Профилактика вовлечения ЦНС является компонентом терапии лейкоза для предотвращения клинически явной лейкемической инфильтрации нервной системы.
В большинстве центров используется сочетание лучевой терапии различными дозами и химиотерапии.
Внутричерепное кровоизлияние редко возникает при остром лимфобластном лейкозе, но является частой причиной смерти при нелимфобластных лейкозах (Yamauchi и Umeda, 1997). Субдуральная гематома или субарахноидальное кровоизлияние обычно являются признаками неблагоприятного прогноза. Тромбоз мозгового синуса (Sebire et al., 2005) может проявляться судорогами и симптомами повышения ВЧД. Черепные невропатии вследствие лейкемической инфильтрации базилярных мягких мозговых оболочек часто вовлекают лицевой, отводящий и слуховой нервы. Эпидуральная компрессия спинного мозга может быть первым проявлением (Pui et al., 1985).

б) Неврологические проявления лимфом:
1. Неходжкинские лимфомы. Несмотря на то, что лимфомы в настоящее время являются распространенной злокачественной опухолью у взрослых (De Angelis, 2001), они до сих пор относительно редко встречаются у детей. Лимфомы могут быть первичными или вторичными после лейкоза (Porto et al., 2005а). Вовлечение ЦНС (Franssila et al., 1987) наблюдалось у 25% из 63 детей с лимфомами на момент установления диагноза Bergeron et al. (1989).
У 10 из этих 16 пациентов отмечались клинические проявления с частым вовлечением черепных нервов, включая снижение слуха и внезапный амавроз; у 7 из них выявлены аномалии СМЖ; у 6 детей развился бессимптомный менингит. Характеристики СМЖ сходны с таковыми при лейкозах. Нейроменингеальные рецидивы возникли у 19 детей (30%). Периферическая нейропатия иногда наблюдается у пациентов с лимфомами и имеет неблагоприятное прогностическое значение. Может происходить вовлечение венозных синусов.
Вовлечение ЦНС гораздо чаще происходит при лимфомах, расположенных на шее, чем при лимфомах с локализацией в брюшной или грудной полости. Наблюдаются различные гистологические типы, включая мелкоклеточные (Kai et al., 1988) и крупноклеточные анапластические лимфомы (Rowsell et al., 2004). По всей видимости, лимфомы Беркитта имеют лучший прогноз, чем другие типы лимфом.
Первичная лимфома ЦНС все больше распознается как осложнение синдрома приобретенного иммунодефицита (СПИД), который, наряду с растущим применением иммуносупрессивной терапии, обусловливает текущую заболеваемость. Первичная лимфома была описана у небольшого числа детей со СПИДом (Rodriguez et al., 1997). У детей со СПИДом лимфомы ЦНС, возможно, более распространены, чем оппортунистические инфекции ЦНС (Epstein et al., 1988). Они могут проявляться как очаговые объемные образования, гиперденсные до введения контраста, с выраженным гомогенным усилением, и дают интенсивный сигнал в режиме Т2 при МРТ.
Возможно появление новых очагов через каждые несколько дней. Двое из трех детей, описанных Epstein et al., имели крупноклеточную лимфому, один — лимфому Беркитта. Эти очаги, которые могут быть ошибочно приняты за воспалительные, очень чувствительны к лечению кортикостероидами, хотя рецидивы в любом случае неизбежны. Лимфомы крайне чувствительны к лучевой терапии, частота ремиссии является высокой (80%).
Влияние лимфом на периферическую нервную систему было рассмотрено в обзоре Hughes et al. (1994).
2. Болезнь Ходжкина. Неврологические осложнения болезни Ходжкина редки у детей (Sapoznik и Kaplan, 1983). Они включают внутримозговые отложения, дающие начало очаговым симптомам, вовлечение базальных базальных менингеальных оболочек с параличами черепных нервов и спинальные экстрадуральные депозиты, которые могут вызвать параплегию. Осложнения обычно сопровождаются отклонениями в составе СМЖ, иногда в форме эозинофильного менингита (Patchell и Perry, 1981). Другой тип вовлечения нервной системы возникает вследствие прямого распространения висцерального поражения на нервную систему, что вызывает компрессию ствола мозга, плечевого сплетения и спинальных корешков (Mallouh, 1989).
У ребенка с болезнью Ходжкина была описана полинейропатия, возникшая не в результате прямой инфильтрации нервов, но, очевидно, в результате непрямого влияния опухоли. Подобные паранеопластические синдромы, распространенные у взрослых, исключительно редко встречаются у детей. У одного ребенка был описан паранеопластический мозжечковый синдром, проявившийся хронической атаксией (Topςu et al., 1992; Hahn et al., 2000). Вторичные опухоли часто возникают через несколько лет после излечения болезни (Lin и Teitell, 2005).
Все лимфомы могут сопровождаться такими же оппортунистическими инфекциями, что и лейкозы.
Инфекционные причины лейкозов - лимфома Беркитта, иммунобластная лимфома, Т-клеточный лейкоз взрослых (ТКЛВ)
В экспериментальной лейкозологии существует много видов вирусов, с помощью которых индуцируют различные формы лейкозов. Известны также вирусы, вызывающие лейкозы у разных видов животных в естественных условиях. Для этих вирусов доказаны вертикальная трансмиссия (передача от матери потомству) и значительное распространение носительства в популяциях животных без признаков заболевания.
Эндемические формы инфекционных гемобластозов
Для большинства эндемических форм гемобластозов человека с определенностью установлено участие в индукции заболевания инфекционных агентов. В отношении некоторых убиквитарных форм существуют лишь более или менее убедительные подозрения. Рабочая группа МАИР в настоящее время причисляет к доказанным этиологическим факторам (группа 1) лимфопролиферативных гемобластозов следующие инфекционные агенты: вирус Эпштейна—Барр (EBV), вирус Т-клеточной лимфомы/лейкоза взрослых (HTLV-1), вирус иммунодефицита человека ВИЧ (HIV) и бактерию Helicobacter pylori.
Носительство EBV распространено очень широко и охватывает до 90 % взрослого населения. Вирус крайне неустойчив во внешней среде, поэтому заражение возможно только при прямом контакте — воздушно-капельным путем через слюну. Возраст, в котором происходит инфицирование, зависит от социальных условий: в развитых странах до трехлетнего возраста инфицируется около 20 % детей, тогда как в развивающихся —до 70 %. EBV обладает высокой тропностью к эпителию ротоглотки, где в нормальных условиях происходит его репродукция, и к В-лимфоцитам, в которых осуществляется бессимптомное носительство в виде кольцевой ДНК (эписома).
Доля содержащих эписому В-лимфоцитов невелика и довольно постоянна — 1 клетка на 10 5 —10 6 . В продуктивной фазе вирусная ДНК в В-клетках приобретает линейную форму.
В экваториальной зоне Африки с EBV связана лимфома Беркитта (ЛБ), которая занимает первое место по распространенности среди гемобластозов у детей на территориях, расположенных до 1550 м над уровнем моря в теплом и влажном климате. Особенно часто заболевание развивается у детей 5—13 лет; в 95 % всех случаев болезнь возникает до 16 лет. У детей моложе 2 лет заболевание практически не встречается. Классическое начало ЛБ: быстро растущая опухоль челюсти или органов брюшной полости у ребенка 5—8 лет. Мальчики преобладают в соотношении (1,7—2,0):1. Неберкиттовские лимфомы в этом регионе исключительно редки.
Морфология лимфомы Беркитта
Повышенное внимание к признакам лимфомы Беркитта (ЛБ) привело к тому, что это новообразование стали обнаруживать в западных странах среди НХЛ детского возраста (до 20 %). Раболеваемость в Африке значительно выше — в среднем 8 на 100 тыс. детей против 0,2 среди белого населения США.
Практически у 100 % больных лимфомой Беркитта (ЛБ) в африканских очагах в клетках опухоли выявляются включения ДНК EBV и определяются высокие титры антител к капсидным и ранним антигенам. Для здоровых детей с антителопродукцией к EBV риск ЛБ повышен в 50—60 раз. Среди спорадических случаев ЛБ носительство ДНК EBV в клетках опухоли много реже — около 30 %. В отличие от общего пула В-клеток ДНК ЕВУ содержится во всех опухолевых клетках ЛБ, причем в моноклоновой форме, что является основным подтверждением этиологической роли вируса. Известна способность EBV к трансформации клетки и индукции бесконечного деления В-лимфоцитов in vitro, тем не менее его роль в патогенезе ЛБ не вполне ясна.
Есть данные, что кофактором в развитии заболевания служит малярийный плазмодий: ареал высокого риска ЛБ совпадает, как правило, с эндемическими регионами малярии. У переселившихся в эти регионы из благополучных районов повышается вероятность возникновения обоих заболеваний. Проведение тотальной лекарственной профилактики малярии приводило к достоверному снижению заболеваемости ЛБ, а после прекращения противомалярийных мероприятий заболеваемость возвращалась на прежний уровень. Эпидемиологические наблюдения позволили построить следующую схему патогенеза ЛБ: малярийная инвазия, хроническая инфекция и, возможно, другие факторы могут служить активаторами пролиферации лимфоцитов.
Контакт с EBV вводит В-лимфоцит в бесконечный цикл деления (нечто близкое к инфекционному мононуклеозу). На следующем этапе случайная хромосомная транслокация способна вывести одну из активированных клеток из-под контроля, и она становится родоначальницей клона.
Анализ 665 случаев лимфомы Беркитта (ЛБ) из регистра Ибадана (Нигерия) показал значимое снижение относительной частоты заболеваний в период 1973—1990 гг. (37,1 % от всех опухолей у детей) по сравнению с 1960—1972 гг. (51,5 %). По всей видимости, это отражает реальное снижение заболеваемости за счет улучшения условий жизни и успешного контроля малярии.
Т-клеточный лейкоз взрослых (ТКЛВ) — заболевание, характерное для юго-восточных областей Японии (Окинава, Киуши, Шикоку), Экваториальной Африки и Карибских островов, Южной Америки и Ближнего Востока. В эндемических очагах до 20 % населения продуцируют антитела к HTLV-1 (среди населения США — 0,025 %). Наиболее распространен HTLV-1 в Африке —до 10 млн инфицированных, всего в мире их 15 — 20 млн. Трансмиссия вируса происходит вертикально (при грудном вскармливании ребенка), а также при гемотрансфузиях. Выявляемость носителей увеличивается с возрастом и достигает пика к 50 годам, причем доля женщин больше. Один случай ТКЛВ в год возникает среди приблизительно 1500 носителей HTLV-1.
В итоге заболевание развивается у 1—4 % носителей через 20— 30 лет после заражения, причем более 90 % заболевших серопозитивны к HTLV-1. Максимальное число заболевших регистрируется в Японии в возрасте около 60 лет, а в Африке — в 40—45 лет. Случаи заболевания детей исключительно редки, частота заболеваний у мужчин и женщин примерно равная. Смертность от ТКЛВ среди инфицированных достигает у мужчин 68, а у женщин 36 на 100 тыс.; среди серонегативных лиц она много ниже. Обследованные в эндемических районах матери заболевших ТКЛВ оказались инфицированными HTLV-1 в 100 %, тогда как матери больных другими формами лимфом — в 30 % случаев.
В Африке выявлено широкое распространение родственного вируса HTLV-2, который подозревается в инициации В-клеточных лейкозов.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Обзор
Светооптическая микроскопия ткани, пораженной диффузной В-клеточной крупноклеточной лимфомой.
Автор
Редакторы

Партнер номинации — компания SkyGen: передовой дистрибьютор продукции для life science на российском рынке.
Генеральный партнер конкурса — международная инновационная биотехнологическая компания BIOCAD.
Введение
Что ж, теперь я хочу пригласить вас в удивительный и сложный мир молекулярного и генетического разнообразия лимфомы. Уверяю вас, что биология рака — как огромный океан: себя можно представить в небольшом батискафе в бездонном морском пространстве; здесь много неизвестного, ты порой не понимаешь, как оценить весь масштаб происходящего, ощущаешь себя мелкой крупицей. Все это пугает и завораживает одновременно. Готовы к погружению?
Такая разная лимфома
Два паттерна экспрессии генов ДВККЛ определяют клинический прогноз
Рисунок 1. Кривые общей выживаемости больных диффузной В-крупноклеточной лимфомой в зависимости от молекулярного подтипа.
Возможно, именно различия в экспрессии генов лежат в основе различного исхода пациентов? Авторы этого же исследования проанализировали общую выживаемость больных, которые получали сопоставимые схемы лечения. Оказалось, что пациенты с GCB-подтипом ДВККЛ имели статистически значимые различия в выживаемости в сравнении с АВС-подтипом (activated B-cell like subtype) ДВККЛ: в среднем общая пятилетняя выживаемость для всех пациентов составила 52%, однако 76% пациентов с GCB-подтипом по достижении пятилетнего рубежа были все еще живы, в то время как только 16% пациентов с ABC-подтипом остались в живых [3]. Исследовательская группа предоставила данные по выживаемости в виде кривых выживаемости Каплана—Мейера (рис. 1). Эти кривые имеют ступенчатый вид и очень часто используются в клинических исследованиях для оценки выживаемости пациентов. В таких графиках каждая ступенька означает наступление неблагоприятного для пациента события: прогрессирование заболевания (при изучении выживаемости без прогрессирования) или летальный исход (при изучении общей выживаемости).
Таким образом, был получен окончательный вывод: диффузная В-клеточная крупноклеточная лимфома представляет собой не одно заболевание, а группу различных по молекулярному патогенезу заболеваний с неодинаковым клиническим прогнозом.
К большому сожалению, даже на сегодняшний день в рутинной онкогематологической практике использование методов анализа экспрессии генов невозможно из-за дороговизны и сложности метода. В качестве альтернативного метода определения молекулярного подтипа ДВККЛ в клинике врачами-патологоанатомами используется окрашивание ткани опухоли в исследовании, основанном на взаимодействии антитела с антигеном — иммуногистохимическая реакция по алгоритму Hans [4], [5]. Данный метод основан на оценке относительного уровня экспрессии всего трех маркеров: CD10, BCL6 и MUM1 (рис. 2). Хоть этот метод более доступен, он не так точен и не позволяет в чистом виде выделить АВС-подтип, поэтому в иммуногистохимическом алгоритме предусмотрено разделение ДВККЛ на GCB- и non-GCB-подтипы.
Рисунок 2. Алгоритм Hans для иммуногистохимической идентификации подтипов.
Молекулярные основы патогенеза ABC- и GCB-подтипов ДВККЛ
Генетические поломки, обуславливающие формирование злокачественных лимфом, возникают на определенном этапе нормального развития В-лимфоцита. Опухолевые В-лимфоциты неходжкинских лимфом имеют биологические особенности нормальных В-лимфоцитов, и они используют эти свойства для собственного выживания и размножения злокачественного клона. Это означает, что функционирование В-клеток лимфом в значительной степени определяется уровнем развития или дифференцировки В-лимфоцитов, из которых опухоль происходит. Обратите внимание на основные последовательные стадии дифференцировки В-лимфоцита (рис. 3), начиная от стволовой кроветворной клетки и заканчивая плазматической клеткой, основная функция которой — секреция защитных антител [6]. Это знание еще пригодится нам в дальнейшем.
Рисунок 3. Последовательные этапы развития В-лимфоцита.
В костном мозге в процессе В-лимфопоэза (формирования В-лимфоцита из стволовой кроветворной клетки) происходит созревание В-клеточного рецептора лимфоцита — будущего специфичного антитела — за счет V(D)J-рекомбинации при участии ферментов RAG1 и RAG2, которые вносят разрывы в двухцепочечную молекулу ДНК, устраняемые простым соединением концов ДНК. Данное молекулярное событие может стать источником формирования хромосомных транслокаций (перестроек) и образования химерных генов, которые в нормальных клетках отсутствуют и гиперэкспрессия которых значима в патогенезе лимфом. Другим потенциальным источником возникновения генетических поломок является герминативный центр лимфатического фолликула, о котором мы уже говорили ранее. Именно здесь происходят события, связанные с модификацией ДНК, которые представляют собой еще один источник мутагенеза — соматическая гипермутация (накопление большего количества мутаций в генах иммуноглобулинов при участии цитозиндезаминазы AID), в процессе которой происходит созревание аффинитета (сродства) В-клеточного рецептора к антигену, и переключение классов иммуноглобулинов. Таким образом, на определенных этапах нормального развития любого В-лимфоцита существуют молекулярные механизмы, которые создают предпосылки к возникновению генетических поломок и, как следствие, — к развитию лимфом. Возникновение той или иной генетической поломки в В-лимфоците носит вероятностный характер.
Рисунок 4. В-клеточная дифференцировка, лимфомагенез и профиль экспрессии генов на различных этапах созревания В-лимфоцита. Злокачественные лимфомы могут возникнуть на разных этапах нормального развития В-лимфоцита. По мере созревания В-лимфоцита его генетическая регуляторная программа переключается с профиля В-лимфоцита герминативного центра на плазмоцитоидный профиль.
Наконец, мы добрались до кульминационной части данного раздела. В случае прогностически более благоприятного варианта — GCB-подтипа ДВККЛ — экспрессионный профиль генов таков, что он соответствует профилю В-лимфоцита герминативного центра, при этом активность молекулярного сигнального пути NF-kB (nuclear factor kappa-B) в таких клетках снижена [7]. Наоборот, в случае клинически неблагоприятного АВС-подтипа ДВККЛ активность NF-kB высока, что может служить одним из механизмов избегания программируемой клеточной гибели — апоптоза. Такие опухолевые клетки экспрессируют маркеры, характерные для плазматических клеток, включая IRF4 и транскрипционный фактор XBP1 — регулятор секреции иммуноглобулинов. Однако при этом полное развитие В-лимфоцитов АВС-подтипа лимфомы в нормальный плазмоцит невозможно из-за блока дифференцировки [7].
Роль активации NF-kB и сигнального пути В-клеточного рецептора в АВС-подтипе ДВККЛ
Как уже было отмечено ранее, успешная стратегия выживания опухолевого В-лимфоцита может объясняться высокой зависимостью от NF-kB для АВС-подтипа ДВККЛ, но не для GCB-подтипа. А также известно о том, что каскадный внутриклеточный сигнальный путь В-клеточного рецептора в случае АВС-подтипа находится в состоянии хронической гиперактивации [7]. Существует довольно несложный способ продемонстрировать, что эти сигнальные пути являются жизненно важными для опухолевых клеток. Для этого достаточно их заблокировать или выключить. На клеточных линиях, являющихся моделями АВС-подтипа ДВККЛ, исследователи Davis R.E. и соавторы (2010) с помощью явления РНК-интерференции продемонстрировали нежизнеспособность опухолевого клона при подавлении экспрессии одного из ключевых посредников сигнала от В-клеточного рецептора — Брутоновской тирозинкиназы [8]. С другой стороны, Lam L.T. и соавторы (2005) успешно подтвердили токсический эффект блокатора IKK, который связан с путем NF-kB в В-лимфоцитах АВС-подтипа [9].
Что, если результаты этих исследований использовать в терапевтических целях? Сложная система внутриклеточных молекул-посредников злокачественного В-лимфоцита АВС-подтипа ДВККЛ может стать интересным объектом терапевтического воздействия (рис. 5). В фармакологической тяжелой артиллерии врача-гематолога уже сегодня имеются блокаторы В-клеточного рецепторного каскада (ибрутиниб — ингибитор Брутоновской тирозинкиназы, дувелисиб — ингибитор PI3K, полатузумаб ведотин — связывает CD79b) и протеасомные ингибиторы (бортезомиб), которые, косвенно воздействуя на NF-kB, предотвращают его проникновение в клеточное ядро и реализацию программы выживания. Более интересной терапевтической мишенью для лечения ДВККЛ могут стать компоненты комплекса CARD11/BCL10/MALT1 (рис. 5). Приблизительно 10% всех АВС-подтипов лимфом имеют активационную мутацию в CARD11, которая обуславливает автономное формирование внутрицитоплазматических белковых агрегатов CARD11/BCL10/MALT1/TRAF6, что приводит к активации IKK, а в последующем — к запуску NF-kB [7]. Причем такая мутация делает независимым сигнальный путь NF-kB от посредников (мессенджеров) сигнала от В-клеточного рецептора, стоящих выше CARD11.
Рисунок 5. Функционирование внутриклеточного сигнального каскада В-клеточного рецептора для нормального В-лимфоцита (А) и в случае В-лимфоцита АВС-подтипа ДВККЛ с мутантным и диким вариантами CARD11 (B).
Повторяющиеся мутационные паттерны ДВККЛ отражают профиль экспрессии генов
Рисунок 6. Секторальные диаграммы, отражающие относительное представительство молекулярного подтипа ДВККЛ на основе профиля экспрессии генов одному из четырех генетических подтипов, и наоборот.
(а) — доля молекулярных подтипов ДВККЛ (АВC-подтип, GCB-подтип и неклассифицируемый) среди генетических вариантов ДВККЛ (MCD, BN2, N1, EZB).
(б) — доля генетических подтипов ДВККЛ (MCD, BN2, N1, EZB) среди молекулярных подтипов ДВККЛ (АВC-подтип, GCB-подтип и неклассифицируемый). (в) — общая доля всех генетических подтипов ДВККЛ среди всех случаев.
Не так давно представления о разнообразии ДВККЛ несколько усложнились. Schmitz et al. (2018) провели секвенирование экзома [10] и транскриптомный анализ RNA-seq 574 образцов ДВККЛ и выявили группы повторяющихся от случая к случаю мутаций в опухолевой ДНК, которые ассоциированы с одним из молекулярных подтипов [11]. Исследовательской группе удалось выделить четыре генетических типа ДВККЛ: MCD (сосуществующие мутации MYD88 и CD79b в опухоли), BN2 (транслокация BCL6 и мутация NOTCH2), N1 (мутация NOTCH1) и EZB (мутация EZH2 и транслокация BCL2) [11]. Только 46,6% случаев ДВККЛ удалось классифицировать по данным генетическим группам; оставшаяся часть лимфом не может быть отнесена ни к одному из подтипов и считается неклассифицированной. Нетрудно заметить (рис. 6), что подавляющее большинство молекулярных подтипов в случае MCD и N1 генетических вариантов соответствуют АВС-подтипу, а EZB вариант — GCB-подтипу [11]. Примечательно, что определение генетических групп, подобно молекулярным группам, имеет клиническое значение с точки зрения прогноза для пациента. Исследование безрецидивной и общей выживаемости пациентов в свете генетических подтипов позволяет сделать вывод о том, что прогностически более неблагоприятными вариантами можно считать N1 и MCD варианты (рис. 7).
Рисунок 7. Кривые безрецидивной (а) и общей (б) выживаемости больных ДВККЛ различных генетических подтипов.
Рисунок 8. Интегральная схема, отражающая взаимоотношение молекулярных подтипов лимфомы на основе профиля экспрессии генов и генетических подтипов, а также потенциальные молекулярные мишени для терапевтических приложений для того или иного генетического типа.
Несмотря на очевидный прогресс в понимании молекулярно-генетического разнообразия и патогенеза ДВККЛ и вопреки многообещающим результатам, полученным при исследовании токсического эффекта таргетных лекарственных препаратов на опухолевые клетки в экспериментальных моделях, на текущий момент в самых последних обновлениях американских клинических онкологических рекомендаций NCCN и в европейских рекомендациях медицинской онкологии ESMO альтернатив режиму R-CHOP в первой линии терапии нет [13].
Такие несоответствия обусловлены тем, что не всегда логичная и стройная идея об эффективности лекарственного препарата, подтвердившаяся в доклинических исследованиях, сможет оправдать себя в клинических исследованиях, в которых принимают участие живые больные люди. Более того, даже наличие статистически значимых различий между экспериментальным лечением и золотым стандартом или плацебо в пользу экспериментального лекарственного препарата не говорит о реальной клинической значимости, выраженной в количестве месяцев или лет жизни, прожитых пациентом в ремиссии в хорошем качестве. Уже имеющиеся результаты крупных клинических исследований II и III фаз по лечению пациентов с впервые выявленной ДВККЛ в попытке улучшить результаты лечения путем добавления ибрутиниба (исследование PHOENIX), леналидомида (исследования ROBUST и E1412) или бортезомиба (исследование REMoDL-B) к стандартному режиму терапии R-CHOP в первой линии, к сожалению, не увенчались успехом [14–17].
Вместо заключения. Зачем изучать такую сложную молекулярную биологию рака?
Если вы уже устали, дойдя до этого момента, из-за всех сложных биологических концепций разнообразия ДВККЛ, то я поспешу вас обрадовать тем, что в этом обзоре освещается лишь только один гистологический вариант лимфомы. Да, современные молекулярно-биологические представления опухолевого роста разнообразны и огромны. Одних внутриклеточных молекулярных посредников, участвующих в каскадных сигналах, существует сколько! А сколько мутаций во всех раковых генах! Весь этот массив информации в голове просто не удержать.
Сегодняшний рынок онкологического биофармацевтического производства представляет совокупность лекарственных препаратов, назначение которых основано на знаниях генетики рака, создавая тем самым возможность прицельного, направленного воздействия на опухоль. Да, да, вы, наверное, уже догадались, что я сейчас говорю именно о персонифицированной или персонализированной медицине в онкологии. Клиницисты сегодня знают довольно немало таких примеров: назначение эрлотиниба пациенту с аденокарциномой легкого при наличии мутации L858R в гене рецептора эпидермального фактора роста EGFR, вемурафениб для лечения распространенной меланомы при обнаруженной мутации в опухоли V600E в гене BRAF, иматиниб как блокатор BCR-ABL при терапии хронического миелолейкоза, мидостаурин в лечении острого миелоидного лейкоза при выявленной мутации в FLT3. Всего этого и многого чего другого просто не было, если бы не развивалась биомедицинская наука. От знаний в молекулярной биологии и генетики выигрывают абсолютно все: ученые удовлетворяют собственный научный интерес и любопытство, врачи принимают участие в клинических исследованиях, развивается фармацевтический рынок, появляются новые профессиональные кадры, а пациенты дольше живут, имея хорошее качество жизни.
Пусть сегодня успехи в выборе клинически эффективного таргетного лекарственного препарата на основе молекулярно-генетических характеристик ДВККЛ не так грандиозны, как хотелось бы, совершенно очевидно одно — дальнейшее усложнение наших представлений в отношении молекулярной генетики опухолей, особенно в условиях расширенной доступности технологий высокопроизводительного секвенирования непременно приведут к революционным терапевтическим подходам в не столь отдаленном будущем. В заключение стоит отметить, что попытка модифицировать схему R-CHOP предпринимается сейчас авторами исследования POLARIX, где они предлагают сравнить режим Pola-R-CHP (полатузумаб ведотин, ритуксимаб, циклофосфамид, доксорубицин, преднизолон) со стандартом, и уже получены результаты: 2-летняя выживаемость без прогрессирования у пациентов из группы лечения Pola-R-CHP составила 76,7% в сравнении с 70,2% у пациентов стандартной терапии R-CHOP. Более того, за последние два года появились возможности лечения рефрактерных или рецидивирующих форм ДВККЛ, такие как биспецифические моноклональные антитела (глофитамаб, мосунетузумаб, эпкоритимаб, одронекстамаб) и CAR-T клеточная терапия.

Обзор
Автор
Редактор
Обратите внимание!
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Исследования в области происхождения злокачественных опухолей, ежегодно уносящих миллионы человеческих жизней, ведутся с XIX века. Работая в области молекулярной вирусологии, Вармус совместно с Дж. М. Бишопом в исследованиях 1970-х гг. сделали открытие, которое по-новому осветило долго остававшуюся дискуссионной проблему этиологии опухолей у человека и животных. Согласно полученным результатам, неконтролируемый рост клеток, образующих опухоль, вызывается не только проникающим в клетку извне онковирусом, но и внутренними процессами в самой клетке. Вармус доказал, что нормальные гены роста клетки вследствие случайных спонтанных мутаций под воздействием химических канцерогенов или, порой, процесса старения могут изменять свою молекулярную структуру и таким образом превращаться в протовирусы онкогенной природы. За открытие клеточного происхождения онкогенных протовирусов Вармус совместно с Дж. М. Бишопом в 1989 г. были удостоены Нобелевской премии по физиологии и медицине [1].
Один из самых распространённых вирусов в человеческой популяции — вирус Эпштейна-Барр (рис. 1) — был открыт и описан в 1964 году двумя английскими вирусологами: Майклом Эпштейном и Ивонной Барр. Вирус Эпштейна-Барр (ВЭБ) — член семейства герпесвирусов [6, 7]. В инфицированных клетках вирусная ДНК, как правило, не встроена в клеточный геном, а находится в ядре в виде замкнутого кольца (эписомы). Биологическое значение интеграции ВЭБ в геном клетки остается неясным. Высказываются предположения, что эписомная ДНК необходима для реализации полноценной репликации ВЭБ, завершающейся формированием вирусных частиц [8].

В отличие от многих других герпесвирусов, вирус Эпштейна-Барр поражает в первую очередь эпителиальные клетки ротовой полости, глотки, миндалин. Здесь он размножается наиболее активно, и поэтому главным путём заражения вирусом являются поцелуи (вот и приехали). Наибольшее количество вирусных частиц находится в клетках эпителия около слюнных желез, и со слюной выделяется большое количество их. Не удивительно, что инфекционный мононуклеоз — самое распространённое заболевание, вызываемое вирусом Эпштейна-Барр, — называют ещё болезнью поцелуев [9].
После первой встречи человека с ВЭБ вирус в незначительном количестве сохраняется в организме хозяина в течение всей жизни. Однако если любой элемент иммунного ответа нарушен, даже незначительное количество ВЭБ-инфицированных клеток может преумножиться колоссально [12].
Инфицированные В-клетки могут значительное время находиться в миндалинах, что позволяет вирусу выделяться во внешнюю среду со слюной. С зараженными клетками ВЭБ распространяется по другим органам. В пораженных вирусом клетках возможно два вида развития: литический, приводящий к разрушению клетки-хозяина, и латентный (клетка заражена, но ничего не выдает нахождения вируса), когда число вирусных копий небольшое и клетка не разрушается. ВЭБ может длительно находиться в В-лимфоцитах, эпителии носоглотки и слюнных железах. Кроме того, он способен проникать и в другие клетки: Т-лимфоциты, NK-клетки, макрофаги, нейтрофилы, эпителиоциты сосудов (рис. 2а, 2б) [13].

Злокачественные лимфомы (например, увеличение лимфоузлов), согласно данным Международного агентства по изучению рака, составляют 3–4% среди всех регистрируемых в мире злокачественных новообразований [14]. Лимфомы делят на две основные группы: лимфома Ходжкина (20–30 % всех лимфом) и неходжкинские лимфомы (около 70%) [14, 15].
Неходжикинские лимфомы — это совокупность новообразований, в возникновении которых принимают участие различные агенты [18]. Первая группа — это вирусы, трансформирующие лимфоциты и другие клетки (ВЭБ, HHV-8). Вторая группа представлена факторами различной природы, вызывающими иммунодефицитные состояния. К таким факторам в первую очередь относится ВИЧ (вирус иммунодефицита человека), вызывающий у инфицированного лица подавление иммунитета в результате истощения пула Т-лимфоцитов CD4+ и возникновение СПИДа. В третью группу входят некоторые инфекции, (например, H. pylori), которые увеличивают риск возникновения лимфом на фоне вызываемой ими хронической стимуляции иммунной системы и постоянной активации лимфоцитов [17].
Исследования последних лет свидетельствуют о том, что в патогенезе ВЭБ-ассоциированных патологий чрезвычайно важную роль играет вредный LMP1 — латентный мембранный белок 1, кодируемый одноименным геном (LMP1). Он обладает свойствами онкобелка и функционирует как постоянно активный псевдорецептор. Он способен изменять В-лимфоциты человека [19].
Есть предположение, что аминокислотные замены, накапливающиеся в LMP1, по-видимому, также вносят свой вклад в возникновение опухолей. Механизм этого процесса окончательно не установлен, но предполагается, что усиленный трансформирующий эффект мутированных LMP1 может представлять важную составляющую этого процесса. При этом показано, что различия в последовательности гена LMP1 могут определять агрессивный географически локализованный генотип ВЭБ [21].
Из известных механизмов действия LMP-2 (второй из братьев семейства LMP), расположенного на противоположном конце линейного генома, упоминается лишь способность этих белков совместно повышать сигнальную трансдукцию в ВЭБ (+) клетках [19].
По данным Харальда цур Хаузена, связь между вирусом и раковым заболеванием считается установленной при определении следующих критериев:
- эпидемиологические доказательства того, что вирусная инфекция является фактором риска для развития специфической опухоли;
- присутствие и сохранение генома вируса в клетках опухоли;
- стимуляция пролиферации клеток после введения генома (или его части) вируса в ткани культуры клеток;
- демонстрация того, что геном возбудителя индуцирует пролиферацию и злокачественный фенотип опухоли [19].
Однако канцерогенность ВЭБ далеко не однозначна. Несмотря на то, что кодируемые вирусом продукты способны вызывать пролиферацию инфицированных клеток, ведущую к возникновению лимфом у больных с иммунодефицитом, эти клинически агрессивные опухоли довольно часто поликлональны и подвергаются регрессии при восстановлении иммунного ответа на ВЭБ. Такие опухоли как лимфома Беркитта (ЛБ) и лимфома Ходжкина (ЛХ) встречаются не только в ВЭБ-ассоциированных, но и в ВЭБ-неассоциированных вариантах, что говорит о том, что патогенез этих новообразований связан не только с ВЭБ. Кроме того, злокачественные клетки больных ЛБ и ЛХ отличаются фенотипически от клеток ЛКЛ, полученных под воздействием ВЭБ in vitro, и не экспрессируют ряд белков, необходимых для трансформирующего роста. Эти находки позволяют предположить, что опухолевые клетки могут возникать и под воздействием факторов невирусного происхождения, а также зависеть от различных усиливающих рост клеток стимулов [17].
Лабораторная диагностика ВЭБ-инфекции базируется на цитологическом исследовании крови или костного мозга, серологических исследованиях и ПЦР. С помощью метода ПЦР можно определить ДНК вируса в плазме до клинических проявлений болезни, а репликация вируса в организме является показанием к противовирусной терапии и критерием эффективности проведенного лечения. Материалом для исследования служат слюна или рото- и носоглоточная слизь, соскоб эпителиальных клеток урогенитального тракта, кровь, спинномозговая жидкость, ткани опухоли и костный мозг. Как у больных ВЭБ, так и у носителей может быть получен положительный результат в ПЦР. Поэтому для их дифференцировки проводится количественный ПЦР-анализ для определения количества копий вирусного генома. У маленьких детей (до 1–3-х лет) по причине недостаточно сформированного иммунитета диагностика по антителам затруднительна, поэтому в данной группе пациентов в помощь приходит именно ПЦР. Однако в силу того, что ПЦР-анализ информативен только при размножении (репликации) вируса, то существует и определенный процент ложноотрицательных результатов (до 30%), связанный именно с отсутствием репликации в момент исследования. При этом важно сопоставление результатов клинических, серологических и молекулярных обследований в определении ВЭБ-инфекции, как причины имеющегося заболевания [1].
Специфическая профилактика (вакцинация) против ВЭБ не разработана, но проводятся клинические испытания. Основной проблемой при разработке вакцины является большое отличие в белковом составе вируса на разных фазах его существования. Впрочем, в настоящее время разрабатывается вакцина, которая содержит рекомбинантный поверхностный антиген gp350. После вакцинации первичная инфекция протекает субклинически, но собственно инфицирование человека не предупреждается. Кроме того, вырабатывающиеся нейтрализующие антитела не влияют на течение различных форм латентной инфекции, в том числе опухолей. Профилактические меры сводятся к укреплению иммунитета, закаливанию детей, мерам предосторожности при появлении больного в окружении, соблюдение правил личной гигиены.
Заключение
Широкое распространение ВЭБ с выраженным трансформирующим потенциалом среди населения планеты и редкого возникновения в инфицированной популяции связанных с этим вирусом опухолей с преимущественной их локализацией в определенных географических регионах позволяет сделать важный вывод. Подобно большинству опухолей иной вирусной природы, в патогенезе ВЭБ-ассоциированных новообразований важную роль играют дополнительные факторы, и одного ВЭБ недостаточно для возникновения опухоли. ВЭБ лишь инициирует пролиферацию инфицированных им клеток, а последующие события влияют на гистопатологический спектр возникающих неоплазий. Одним из важнейших факторов, в значительной степени определяющих возникновение ВЭБ-ассоциированных опухолей, служит выраженная иммуносупрессия (врожденная, ятрогенная или индуцированная любой вирусной инфекцией, и в первую очередь ВИЧ), приводящая к утрате функции иммунного распознавания клеток, инфицированных ВЭБ.
Таким образом, несмотря на многолетнее изучение связи ВЭБ с опухолями человека, вопрос о роли вируса в их возникновении до конца не изучен. Раскрытие механизма злокачественной трансформации вирусом, персистирующим в латентном состоянии более чем у 90% населения планеты, — задача чрезвычайно сложная. Однако технические достижения последних лет, существенно повысившие специфичность исследований, позволяют надеяться, что детали ВЭБ-ассоциированного канцерогенеза будут выяснены.
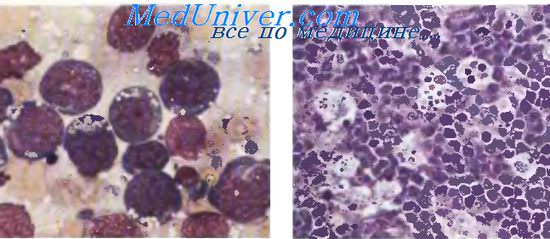
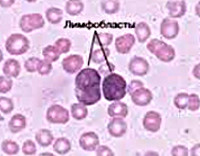
Острый лимфобластный лейкоз – злокачественное поражение системы кроветворения, сопровождающееся неконтролируемым увеличением количества лимфобластов. Проявляется анемией, симптомами интоксикации, увеличением лимфоузлов, печени и селезенки, повышенной кровоточивостью и дыхательными расстройствами. Из-за снижения иммунитета при остром лимфобластном лейкозе часто развиваются инфекционные заболевания. Возможно поражение ЦНС. Диагноз выставляется на основании клинических симптомов и данных лабораторных исследований. Лечение – химиотерапия, радиотерапия, пересадка костного мозга.
МКБ-10
Общие сведения
В соответствии с классификацией ВОЗ различают четыре типа ОЛЛ: пре-пре-В-клеточный, пре-В-клеточный, В-клеточный и Т-клеточный. В-клеточные острые лимфобластные лейкозы составляют 80-85% от общего количества случаев заболевания. Первый пик заболеваемости приходится на возраст 3 года. В последующем вероятность развития ОЛЛ повышается после 60 лет. Т-клеточный лейкоз составляет 15-20% от общего количества случаев болезни. Пик заболеваемости приходится на возраст 15 лет.
Причины острого лимфобластного лейкоза
Непосредственной причиной острого лимфобластного лейкоза является образование злокачественного клона – группы клеток, обладающих способностью к неконтролируемому размножению. Клон образуется в результате хромосомных аберраций: транслокации (обмена участками между двумя хромосомами), делеции (утраты участка хромосомы), инверсии (переворота участка хромосомы) или амплификации (образования дополнительных копий участка хромосомы). Предполагается, что генетические нарушения, вызывающие развитие острого лимфобластного лейкоза, возникают еще во внутриутробном периоде, однако для завершения процесса формирования злокачественного клона нередко требуются дополнительные внешние обстоятельства.
В числе факторов риска возникновения острого лимфобластного лейкоза обычно в первую очередь указывают лучевые воздействия: проживание в зоне с повышенным уровнем ионизирующей радиации, радиотерапию при лечении других онкологических заболеваний, многочисленные рентгенологические исследования, в том числе во внутриутробном периоде. Уровень связи, а также доказанность наличия зависимости между различными лучевыми воздействиями и развитием острого лимфобластного лейкоза сильно различаются.
Так, взаимосвязь между лейкозами и лучевой терапией в наши дни считается доказанной. Риск возникновения острого лимфобластного лейкоза после радиотерапии составляет 10%. У 85% пациентов болезнь диагностируется в течение 10 лет после окончания курса лучевой терапии. Связь между рентгенологическими исследованиями и развитием острого лимфобластного лейкоза в настоящее время остается на уровне предположений. Достоверных статистических данных, подтверждающих эту теорию, пока не существует.
Вероятность развития острого лимфобластного лейкоза повышается при контакте матери с некоторыми токсическими веществами в период гестации, при некоторых генетических аномалиях (анемии Фанкони, синдроме Дауна, синдроме Швахмана, синдроме Клайнфельтера, синдроме Вискотта-Олдрича, нейрофиброматозе, целиакии, наследственно обусловленных иммунных нарушениях), наличии онкологических заболеваний в семейном анамнезе и приеме цитостатиков. Некоторые специалисты отмечают возможное негативное влияние курения.
Симптомы острого лимфобластного лейкоза
Болезнь развивается стремительно. К моменту постановки диагноза суммарная масса лимфобластов в организме может составлять 3-4% от общей массы тела, что обусловлено бурной пролиферацией клеток злокачественного клона на протяжении 1-3 предыдущих месяцев. В течение недели количество клеток увеличивается примерно вдвое. Различают несколько синдромов, характерных для острого лимфобластного лейкоза: интоксикационный, гиперпластический, анемический, геморрагический, инфекционный.
Интоксикационный синдром включает в себя слабость, утомляемость, лихорадку и потерю веса. Повышение температуры может провоцироваться как основным заболеванием, так и инфекционными осложнениями, которые особенно часто развиваются при наличии нейтропении. Гиперпластический синдром при остром лимфобластном лейкозе проявляется увеличением лимфоузлов, печени и селезенки (в результате лейкемической инфильтрации паренхимы органов). При увеличении паренхиматозных органов могут появляться боли в животе. Увеличение объема костного мозга, инфильтрация надкостницы и тканей суставных капсул могут становиться причиной ломящих костно-суставных болей.
О наличии анемического синдрома свидетельствуют слабость, головокружения, бледность кожи и учащение сердечных сокращений. Причиной развития геморрагического синдрома при остром лимфобластном лейкозе становятся тромбоцитопения и тромбозы мелких сосудов. На коже и слизистых выявляются петехии и экхимозы. При ушибах легко возникают обширные подкожные кровоизлияния. Наблюдаются повышенная кровоточивость из ран и царапин, кровоизлияния в сетчатку, десневые и носовые кровотечения. У некоторых больных острым лимфобластным лейкозом возникают желудочно-кишечные кровотечения, сопровождающиеся кровавой рвотой и дегтеобразным стулом.
Иммунные нарушения при остром лимфобластном лейкозе проявляются частым инфицированием ран, царапин и следов от уколов. Могут развиваться различные бактериальные, вирусные и грибковые инфекции. При увеличении лимфатических узлов средостения отмечаются нарушения дыхания, обусловленные уменьшением объема легких. Дыхательная недостаточность чаще обнаруживается при Т-клеточном остром лимфобластном лейкозе. Нейролейкозы, спровоцированные инфильтрацией оболочек спинного и головного мозга, чаще отмечаются во время рецидивов.
При вовлечении ЦНС выявляются положительные менингеальные симптомы и признаки повышения внутричерепного давления (отек дисков зрительных нервов, головная боль, тошнота и рвота). Иногда поражение ЦНС при остром лимфобластном лейкозе протекает бессимптомно и диагностируется только после исследования цереброспинальной жидкости. У 5-30% мальчиков появляются инфильтраты в яичках. У пациентов обоих полов на коже и слизистых оболочках могут возникать багрово-синюшные инфильтраты (лейкемиды). В редких случаях наблюдаются выпотной перикардит и нарушения функции почек. Описаны случаи поражений кишечника.
С учетом особенностей клинической симптоматики можно выделить четыре периода развития острого лимфобластного лейкоза: начальный, разгара, ремиссии, терминальный. Продолжительность начального периода составляет 1-3 месяца. Преобладает неспецифическая симптоматика: вялость, утомляемость, ухудшение аппетита, субфебрилитет и нарастающая бледность кожи. Возможны головные боли, боли в животе, костях и суставах. В период разгара острого лимфобластного лейкоза выявляются все перечисленные выше характерные синдромы. В период ремиссии проявления болезни исчезают. Терминальный период характеризуется прогрессирующим ухудшением состояния больного и завершается летальным исходом.
Диагностика острого лимфобластного лейкоза
Диагноз выставляют с учетом клинических признаков, результатов анализа периферической крови и данных миелограммы. В периферической крови пациентов с острым лимфобластным лейкозом выявляются анемия, тромбоцитопения, повышение СОЭ и изменение количества лейкоцитов (обычно – лейкоцитоз). Лимфобласты составляют 15-20 и более процентов от общего количества лейкоцитов. Количество нейтрофилов снижено. В миелограмме преобладают бластные клетки, определяется выраженное угнетение эритроидного, нейтрофильного и тромбоцитарного ростка.
В программу обследования при остром лимфобластном лейкозе входят люмбальная пункция (для исключения нейролейкоза), УЗИ органов брюшной полости (для оценки состояния паренхиматозных органов и лимфатических узлов), рентгенография грудной клетки (для обнаружения увеличенных лимфоузлов средостения) и биохимический анализ крови (для выявления нарушений функции печени и почек). Дифференциальный диагноз острого лимфобластного лейкоза проводят с другими лейкозами, отравлениями, состояниями при тяжелых инфекционных заболеваниях, инфекционным лимфоцитозом и инфекционным мононуклеозом.
Лечение и прогноз при остром лимфобластном лейкозе
Основой терапии являются химиопрепараты. Выделяют два этапа лечения ОЛЛ: этап интенсивной терапии и этап поддерживающей терапии. Этап интенсивной терапии острого лимфобластного лейкоза включает в себя две фазы и длится около полугода. В первой фазе осуществляют внутривенную полихимиотерапию для достижения ремиссии. О состоянии ремиссии свидетельствуют нормализация кроветворения, наличие не более 5% бластов в костном мозге и отсутствие бластов в периферической крови. Во второй фазе проводят мероприятия для продления ремиссии, замедления или прекращения пролиферации клеток злокачественного клона. Введение препаратов также осуществляют внутривенно.
Продолжительность этапа поддерживающей терапии при остром лимфобластном лейкозе составляет около 2 лет. В этот период больного выписывают на амбулаторное лечение, назначают препараты для перорального приема, осуществляют регулярные обследования для контроля над состоянием костного мозга и периферической крови. План лечения острого лимфобластного лейкоза составляют индивидуально с учетом уровня риска у конкретного больного. Наряду с химиотерапией используют иммунохимиотерапию, радиотерапию и другие методики. При низкой эффективности лечения и высоком риске развития рецидивов осуществляют трансплантацию костного мозга. Средняя пятилетняя выживаемость при В-клеточном остром лимфобластном лейкозе в детском возрасте составляет 80-85%, во взрослом – 35-40%. При Т-лимфобластном лейкозе прогноз менее благоприятен.
Читайте также: