
Математическая модель вирус гриппа
Обновлено: 24.04.2024
Эпидемии издавна угрожали человечеству, и только в ХХ веке были разработаны эффективные средства борьбы с инфекциями. К числу этих средств принадлежат и системы дифференциальных уравнений — математика помогает моделировать распространение эпидемий и помогает понять, как следует с ними бороться. Это наш третий материал о самых интересных дифференциальных уравнениях и о том, где и как они применяются (предыдущие материалы можно прочитать здесь и здесь). Если вы читаете нас с телефона, переключайте страницу на десктопную версию, так вы сможете увидеть интерактивный график целиком.
В XXI веке мир уже успел столкнуться с эпидемией птичьего гриппа в Юго-Восточной Азии (в 2013 году) и вспышкой заболеваний лихорадкой Эбола в Африке (2015). Но в истории человечества бывали и куда более масштабные эпидемии.
В 551-580 годах нашей эры в Восточной Римской империи разразилась первая задокументированная пандемия чумы, получившей название Юстиниановой, в результате которой погибло около 100 миллионов человек (по другим данным, жертв могло быть значительно меньше). Спустя еще 800 лет в Евразию и Северную Африку пришла Черная смерть — пандемия чумы, сразившая от трети до половины тогдашнего населения этих регионов.
В результате Первой мировой войны, вызвавшей перемещение большого количества людей, в 1918 году распространился испанский грипп, охвативший более 500 миллионов человек и погубивший каждого десятого заболевшего. Эта пандемия стала самой масштабной за всю историю человеческой цивилизации, коснувшись до 30 процентов населения Земли.
В медицинской классификации эпидемией называют прогрессирующее распространение инфекционного заболевания на уровне выше среднего на данной территории. В случае распространения эпидемии на большие территории или территории многих стран говорят о пандемии.
Для эпидемии среди животных применяется термин эпизоотия, а среди растений — эпифития. Этим явлениям ученые также уделяют большое внимание, поскольку они, в свою очередь, помогают понять механизм распространения инфекций.
Изучение механизмов развития и распространения эпидемий является важным способом борьбы с заболеваниями наряду с поиском новых лекарств, вакцинацией и профилактическими мерами. На помощь медикам пришли математики — для этого им пришлось объединить дифференциальные уравнения и теорию вероятности.
Первую попытку использовать математический аппарат для исследования механизмов распространения заболеваний предпринял Даниил Бернулли, ранее открывший первые законы гидродинамики. Следующий шаг сделал Уильям Фарр, применивший в 1840 году нормальное распределение к анализу смертности от оспы.
В рамках этой модели с помощью систем дифференциальных уравнений (при условии непрерывности времени и большой популяции) или разностных уравнений (при дискретном времени и ограниченной популяции) описывается динамика распространения заболевания.
Модель SIR
SIR–модель получила заслуженную популярность в силу простоты построения и использования. Ее применение позволяет точно моделировать эпидемии гриппа и других заболеваний в больших городах, вводить новые параметры и анализировать разные сценарии.
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.

Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.

Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Белки: как получают, хранят и отображают информацию об их структуре?
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.

Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.
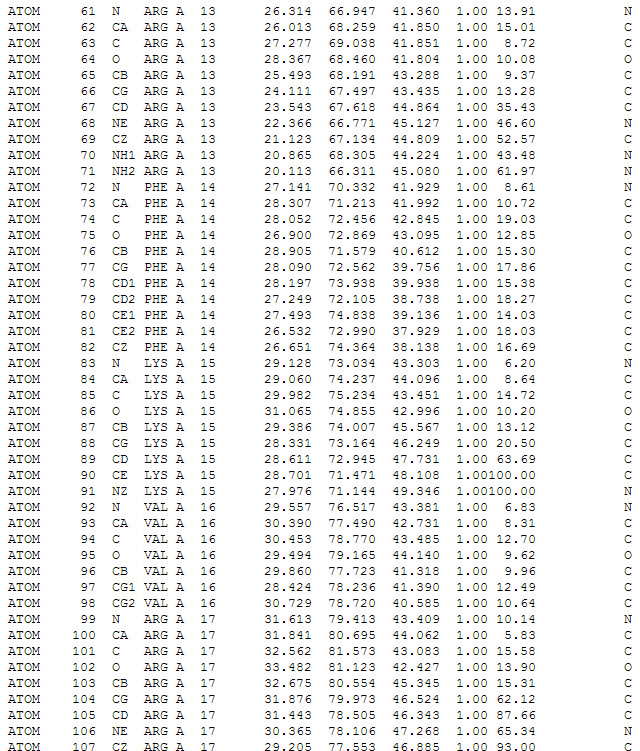
Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.

Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:

Изображения молекулы иммуноглобулина G.
Молекулярное моделирование

Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Докинг
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Посттрансляционные модификации
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Молекулярная динамика и оптимизация структур
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.

В 2020 году мир как будто забыл о сезонном гриппе благодаря пандемии коронавируса. Однако грипп никуда не делся. Некоторые эпидемиологи ожидают вспышку заболеваемости уже в сезоне 2021–2022. Грипп может проявиться агрессивнее, потому что наша иммунная система, привыкшая к ежегодной эпидемии, ослабила хватку. При этом, говорят эксперты, из-за дефицита информации в сезоне 2020–2021 труднее спрогнозировать ситуацию и определить, какие штаммы будут распространяться.
Действительно ли так сложно прогнозировать эпидемию? Ведь мы с гриппом сосуществуем не одно десятилетие, в отличие от COVID-19 и, должно быть, все о нем знаем. Предлагаю разобраться — с минимумом уравнений, без которых никуда, и сложных терминов.
Что такое эпидемия с научной точки зрения
Великая чума в Лондоне, унесшая в 1665–1666 годах жизни 100 тысяч горожан (20 % населения), — эпидемия. Вспышка гриппа в 1978 году в закрытой школе для мальчиков на севере Англии, в результате которой переболело 512 из 763 учеников, — тоже эпидемия. Относительно недавний случай во Франции, когда 7 гостей на свадебной вечеринке заразились вирусным гепатитом E, съев традиционную корсиканскую колбасу из свиной печени (фигателлу), также считается эпидемией.
Огромная доля эпидемий проходит незамеченными для большинства из нас. По данным ВОЗ, с 2011 по 2017 год в мире фиксировалось от 164 до 213 эпидемий: чума, холера, лихорадка Денге, вирус Зика и другие.

Одним словом, эпидемий так много, что логично подходить к ним системно, привлекая математику, чтобы сгладить их последствия.
Вирус гриппа и первая сложность для моделирования
Грипп — вирус, для которого естественным резервуаром являются животные: птицы, свиньи, лошади, люди. Существует четыре типа гриппа: A, B, C и D, из которых у людей встречаются первые три, при этом тип C выявляется редко и обычно безобиден. Самый опасный, приводящий к пандемиям, — вирус группы A. Он подразделяется на подтипы в зависимости от сочетания белков (антигенов) на его поверхности, которые обеспечивают проникновение в клетку человека: гемагглютинина (HA) и нейраминидазы (NA).
Важно отметить, что грипп — РНК-вирус, что означает высокую вероятность мутаций. В процессе репликации (копирования) РНК при проникновении в клетку происходит больше количество ошибок из-за изменений в структуре гемагглютинина и нейраминидазы. Грубо говоря, в клетке собирается не тот вирус, который ожидает встретить организм. Это новый штамм, к которому нет иммунитета, даже если организм перенес грипп месяц назад. Запомним эту важную для моделирования особенность.
Самая популярная модель и другие сложности для моделирования
При моделировании, как правило, смотрят на следующие характеристики, которые помогают рассчитать нагрузку на систему здравоохранения:
время наступления пика эпидемии;
количество людей, которые переболеют за время эпидемии.
Для ответа на эти вопросы в 1927 году шотландские ученые Андерсон Кермак и Уильям МакКендрик предложили SIR-модель, которая до сих пор считается базовой. Согласно ей, вся популяция разделена на три группы: восприимчивых к инфекции (Susceptible), инфицированных (Infectious) и переболевших, приобретших иммунитет (Recovered).
Сразу можно сказать, что для гриппа остановка на Recovered не всегда работает. Допустим, человек вылечился, приобрел антитела и, уверенный в своей неуязвимости, поехал в офис на метро. Через два дня снова лежит дома в постели с температурой, головной болью и прочими признаками гриппа. Почему? Потому что на территории циркулируют два разных штамма, отличающиеся комбинацией гемагглютинина и нейраминидазы. И один их них иммунная система вовремя не распознала. Плюс организм ослаб после борьбы с предыдущим штаммом — возможно, это обстоятельство приведет к более тяжелому течению болезни, вызову скорой помощи и госпитализации.
Модель SIR описывается системой из трех дифференциальных уравнений:

С точки зрения математики то, что подкрашено красным, означает, что мы имеем дело с нелинейностью. Мы не сможем найти решение аналитически, составляя формулы через известные нам функции. Приходится применять численное решение. Коэффициенты, которые здесь фигурируют, определяют скорость перехода из одной группы в другую: β — скорость перехода из здоровых в заболевшие, γ — из заболевших в выздоровевшие.
В конечном итоге мы получим тождество:

S(0) — количество людей, которые могут заболеть до начала эпидемии. И это же индекс репродукции R0, хорошо известный всем, кто следил за развитием пандемии COVID-19. Он означает, сколько человек может заразить один заболевший. При R0 больше единицы развивается эпидемия. Если индекс меньше единицы, эпидемия затухает.
SIR-модель подразумевает, что люди составляют непрерывную, однородную среду. Как газ или жидкость. Популяция считается постоянной, не учитывается убыль населения — естественная или от вируса — и прибыль. На практике это приводит к серьезной погрешности в расчетах.
Помните пример с гриппом в закрытой школе для мальчиков? Даже если с помощью математики смоделировать ситуацию в школе с доверительным интервалом (95 %), получится, что переболели примерно 750 учеников. Но мы точно знаем, что заболевших было 512. R0 примерно равен 16, а на самом деле был 1,69. Если бы мы так же в лоб моделировали ситуацию не для одной школы, а, скажем, для города или страны, наши прогнозы вызвали бы тысячи вопросов. Как же быть?
Важно понимать, что грипп не развивается молниеносно. Есть латентный период и инкубационный период. От начала заражения до появления клинических симптомов — в среднем 2 дня (у COVID-19, как вы знаете, до 14 дней). При этом инкубационный период частично пересекается с периодом инфицирования: больной еще не знает, что болен, но уже заражает окружающих.

Для учета зараженных в инкубационном периоде в SIR-модель вводят группу E (Exposed). Модифицированная модель SEIR — это новая система уравнений, новый коэффициент, но R0 по-прежнему получается 16.
Подобных вариаций SIR-модели много. Если, например, принимать в расчет количество умерших, но без карантина, получим модель SEIRD и т. д. Однако что бы мы ни предпринимали, SIR-моделирование не дает приемлемой точности. Дело в том, что любая модификация модели является детерменистской: для нее не существует случайных причин. Грипп же предполагает массу случайностей на каждом этапе, от прикрепления вируса к клетке до прохождения многочисленных ловушек иммунитета. Закрепиться в нашем организме — квест для каждого вириона.
Модели, которые учитывают случайности
Если количество уязвимых к инфекции и заболевших — случайные величины, модель называют стохастической. Она пытается ответить на вопрос: если на k-неделе x — это количество здоровых, то какая вероятность, что на неделе k+1 х будет означать количество заболевших? И кроме того — какая вероятность избежать инфекции (u)?
Когда u зависит от числа заболевших (в том смысле, что чем больше заболевших, тем меньше вероятность избежать заболевания), мы получаем модель Рида-Фроста. Если же предполагаем, что вероятность заболеть не зависит от количества заболевших, получаем модель Гринвуда.

Это уже более гибкие модели, не непрерывные — их еще называют ветвящимися (branching model). Число выздоровевших убывает, вероятность заболеть в ближайшую неделю меняется в зависимости от ситуации на предыдущей неделе. Стохастические модели получили широкое распространение в 1970-е, постоянно развивались и сейчас в целом имеют неплохое согласие с наблюдениями различных инфекций.
Что еще можно учесть? Например, то, что люди по-разному социализированы. Кто-то пять дней в неделю общается с десятками коллег и постоянно встречается с друзьями, у кого-то друзей нет и работа из дома. Существует вероятность, что у них разные шансы заболеть гриппом, поэтому можно рассматривать распространение инфекции на социальном графе. Выглядит он примерно так:

С таким моделированием тоже есть проблемы, потому что на практике невозможно построить граф, фиксируя все социальные контакты.
Что еще нужно учитывать?
Вернемся к процессу заражения. При дыхании, чихании и кашле инфицированного в воздух попадают частицы, которые делят по размеру: больше 5 микрометров (Droplet, капля) и меньше 5 микрометров (Airborne, зародыш капли). Если кашель генерирует примерно 10 тысяч частиц, чихание — миллион. Поэтому следует сторониться прежде всего тех, кто чихает.
Частиц меньше 5 микрометров большинство. Они почти беспрепятственно проходят через верхние дыхательные пути и попадают в легкие. Там уже нет ресничкового эпителия, густо покрытого слизью, который служил серьезным барьером для частиц побольше. Зато есть альвеолярные макрофаги нейтрофилы, готовые атаковать любой патоген, правда, не всегда эффективно. Зародыши капли легко переносятся по воздуху и долго оседают, в 4 раза медленнее частиц размером 10 микрометров.
Серьезное значение для распространения вируса имеет скорость испарения частиц. Чтобы испариться наполовину, нужно от 0,01 до 10 секунд. Разница напрямую связана с влажностью: чем ниже влажность, тем быстрее испаряется частица. Больной в лифте чихнул, влажность низкая — частица, не успев осесть, испарилась до размера зародыша и продолжает парить в воздухе, пока не попадет в нос здоровому человеку.
Теперь вспомним, когда влажность в помещениях минимальна. В Германии, например, в феврале и декабре — на пике отопительного сезона. Вот почему у гриппа выраженная сезонность (у туберкулеза, скажем, такой сезонности нет). Таким образом, знание динамики аэрозольных частиц позволяет объяснить, почему эпидемии возникают в определенное время года. Во-вторых, помогает оценить риск заражения.
Вместо заключения
Такие предсказания, несмотря на все погрешности, необходимы. С помощью SIR-модели можно определить, например, что эпидемия начнется в первой декаде декабря, продлится три недели, заболеет 1 миллион человек, в пике будет 100 тысяч случаев инфицирования. Это позволит системе здравоохранения подготовиться, приготовить нужное количество лекарств и мест в больницах. Затем имеет смысл применять вероятностные модели, уточняя прогнозы: учитывать вакцинированных, вероятность заразиться каждой группе в популяции и т. д. Пока что это оптимальная практика, помогающая справляться с эпидемиями вирусов, которые мы неплохо знаем.

Мораль простая: полезно мыть руки, проветривать помещения, включать увлажнитель, но без вакцинации победить эпидемию гриппа нельзя. И неважно, какую модель использовать для предсказаний.
Если вам интересно, как математика и машинное обучение помогают бороться с фейковыми новостями, слухами и теориями заговора во время эпидемий, рекомендуем доклад Преслава Накова на бесплатной онлайн-конференции IT NonStop (18–20 ноября 2021). Преслав Наков — главный научный сотрудник Катарского исследовательского института вычислительной техники, HBKU. Он возглавляет мегапроект Tanbih, разработанный в сотрудничестве с MIT, с помощью которого, в частности, выявляют фейки о COVID-19.
Всего в программе конференции — более 40 докладов и воркшопов специалистов из Microsoft, AWS, Ocado, Codete, Ciklum, Eleks, SoftServe, Toloka, Yandex, DataArt и других компаний.
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.
Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.
Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Белки: как получают, хранят и отображают информацию об их структуре?
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.
Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.
Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.
Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:
Изображения молекулы иммуноглобулина G.
Молекулярное моделирование
Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Докинг
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Посттрансляционные модификации
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Молекулярная динамика и оптимизация структур
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.
Читайте также: