
Модель коронавируса своими руками
Обновлено: 19.04.2024
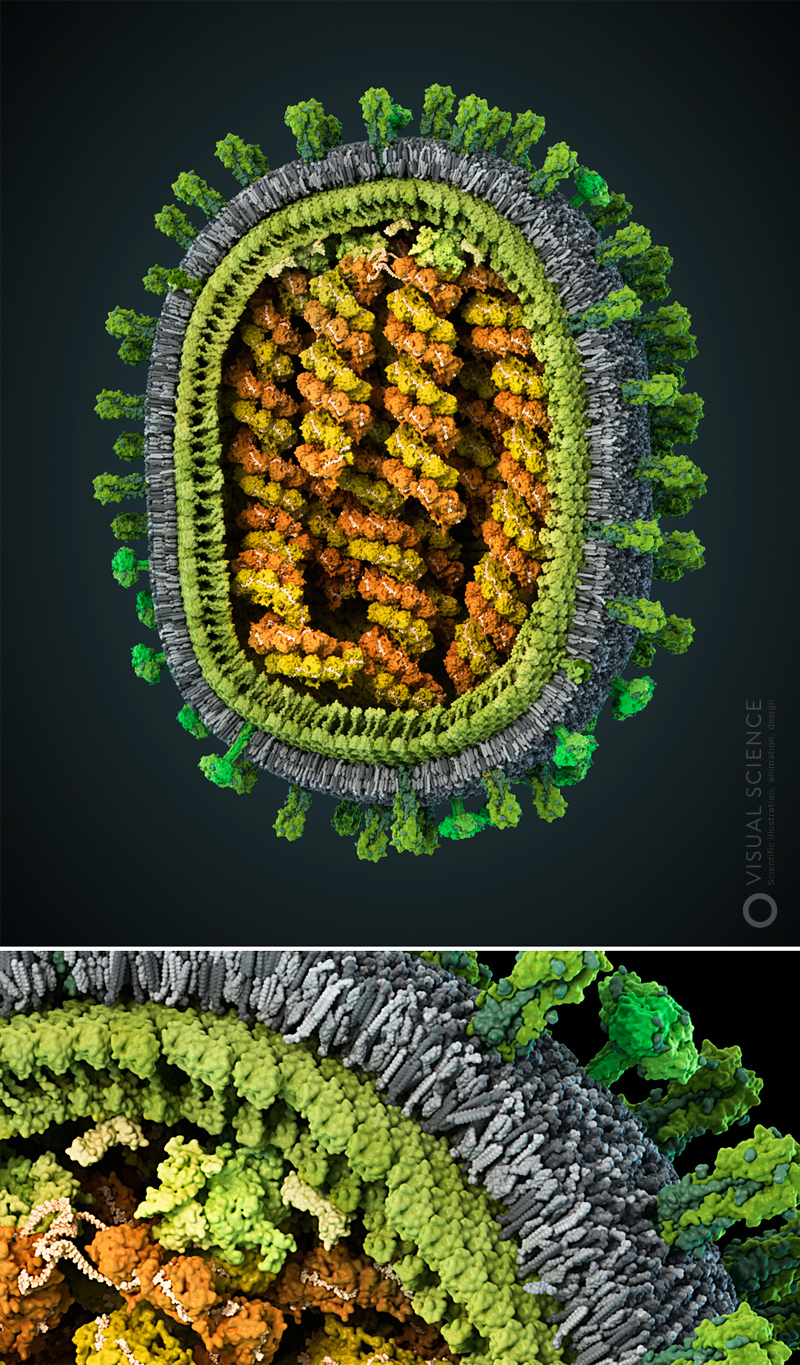
В студии биомедицинской визуализации Visual Science создана наиболее детальная и научно достоверная трехмерная модель вируса SARS-CoV-2 в атомном разрешении. Модель основана на современных научных данных о строении коронавирусов, а также комментариях ведущих вирусологов, занимающихся изучением этой группы вирусов. Это наиболее достоверная модель из представленных на данный момент. При ее создании в Visual Science были использованы подходы и методы, применяемые в фундаментальных и прикладных научных исследованиях.
Модель SARS-CoV-2 является частью некоммерческого проекта студии Visual Science Зоопарк вирусов, в рамках которого уже созданы модели ВИЧ, гриппа A/H1N1, вируса Эбола, вируса папилломы человека и вируса Зика.
Модели и визуализации, созданные в рамках Зоопарка вирусов, отмечены призами журнала Science и Национального научного фонда США, и были опубликованы в ведущих медиа, среди которых Science, Nature Medicine, The New York Times, The Washington Post, Scientific American, Wired UK, Der Spiegel, Stern, National Geographic, GEO и другие.
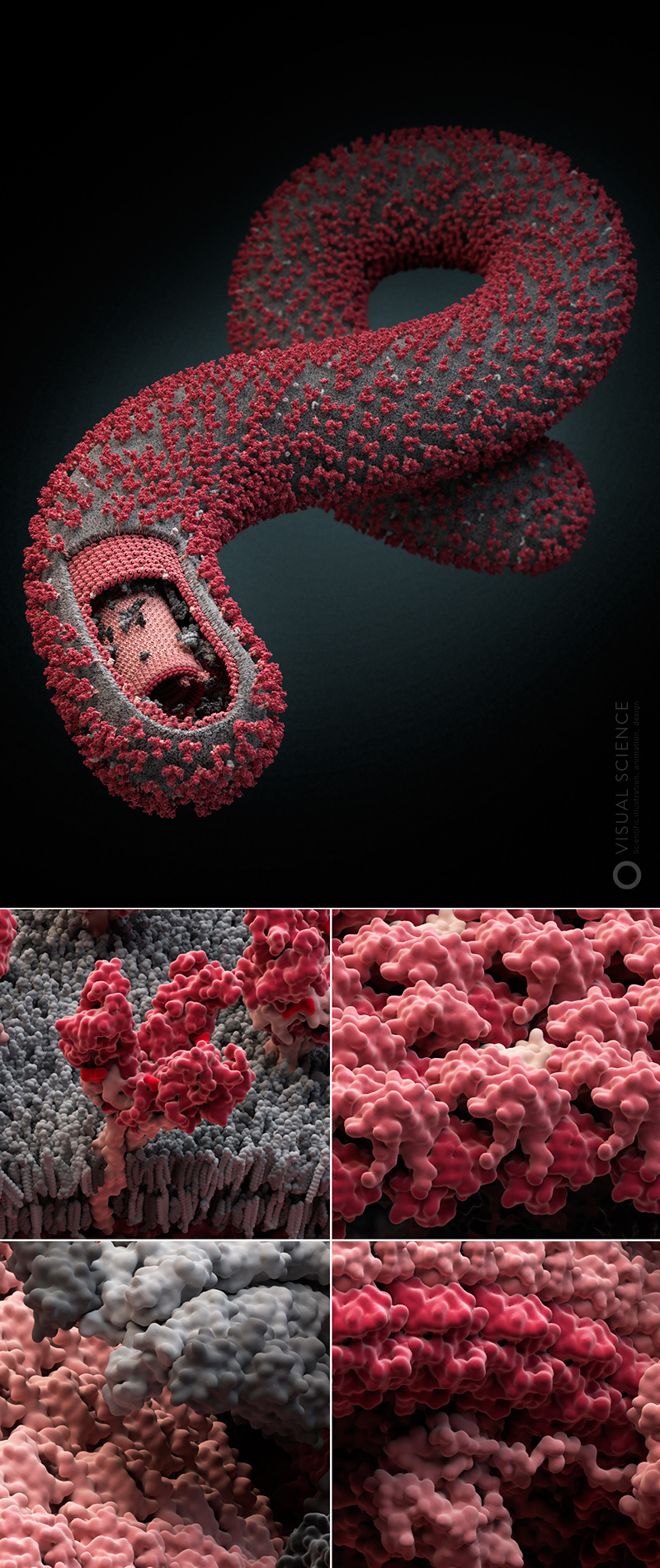
В свойственном им завораживающем стиле, талантливые иллюстраторы из Visual Science создали модель вирусной частицы SARS-CoV-2. В мельчайших деталях она демонстрирует зрелую частицу с поверхностными белками, встроенными в мембрану. Вид со срезом показывает нуклеокапсид внутри частицы. Эти великолепные изображения позволяют лучше понять организацию вируса, а для неспециалистов, сделать вирус, инфецирующий миллионы людей, практически осязаемым.
— Винсент Раканиелло, Хиггинсовский профессор Департамента Микробиологии и Иммунологии Колумбийского университета, Нью-Йорк.
Отличная работа с поверхностными белками и иллюстрациями! По-настоящему хорошо сделано. Поверхностные выросты показаны настолько аккуратно, насколько это в принципе возможно, опираясь на современные данные. Изображения великолепны.
— Джейсон МакЛиллан, профессор, Департамент биомолекулярных наук Университета Техаса в Остине
Ого, это по-настоящему здорово. Мне нравится вариант, когда частица открыта, и видно РНП — новая любимая модель SARS-CoV-2.
— Бенджамин Ньюман, профессор, глава Отдела биологических наук Техасского механико-агрикультурного университета Тексаркана
Модель была построена на основании предположений, происходящих из всех доступных на данный момент сведений. Это хорошая модель. Тем не менее, надо иметь в виду, что исходные структуры С-концевых участков нуклеокапсида были получены без РНК. Присоединение РНК, вероятно, еще компактизует структуру, благодаря сильным РНК-белковым взаимодействиям.
— Профессор Тай-хуанг Хуанг, заслуженный научный сотрудник Отделения структурной биологии Института биомедицинских наук Академии Синика, Нанканг, Тайпей
Вирус SARS-CoV-2
В конце декабря 2019 года подразделение Всемирной организации здравоохранения (ВОЗ) в Китае зарегистрировало вспышку пневмонии неизвестного происхождения. В начале января 2020 года был определен возбудитель заболевания — коронавирус, родственный возбудителям тяжелого острого респираторного синдрома (SARS) и ближневосточного респираторного синдрома (MERS). Позднее разновидность вируса была названа SARS-CoV-2, а вызываемое им заболевание — COVID-19 (от английского Corona Virus Disease). 11 марта 2020 года ВОЗ официально объявила о начале пандемии нового заболевания [1].
Что такое коронавирусы?
Вирусы — это крайне разнообразная группа агентов. Некоторые из них содержат только белки и генетический материал (например, вирусы обычной простуды или вирус папилломы), а у некоторых, как у гриппа или ВИЧ, есть оболочка, которую вирус похищает из клетки хозяина. Оболочка состоит из двойного слоя липидов, в который встраиваются белки вируса. У коронавирусов такая оболочка есть.
Геномы вирусов, в отличие от геномов бактерий или более сложных организмов, могут состоять как из ДНК, так и из РНК. У коронавирусов генетический материал представлен РНК и имеет самую большую длину из всех вирусов со сходной организацией — около 30000 нуклеотидов (структурных элементов, последовательность которых и несет информацию о строении белков вируса) [6]. Для сравнения, геном вируса гриппа, также представленный одноцепочечной РНК, хотя и разделенной на фрагменты, суммарно примерно в 2 раза короче [7]. Обычно, у вирусов более длинный геном означает возможность кодировать больше белков и, как следствие, вступать в более сложные взаимоотношения с клеточными системами защиты.
Среди коронавирусов ученые выделяют 4 крупные группы, названные буквами греческого алфавита — от альфа до дельта. Альфа- и бетакоронавирусы заражают млекопитающих, тогда как гамма- и дельтакоронавирусы заражают преимущественно птиц и рыб [8,9].
Коронавирусы животных, при этом могут причинять существенный экономический ущерб. Так, в 2016 году, вирус, вызывающий острую диарею у свиней, во время вспышки погубил порядка 24000 поросят [10].
До 2019 года было известно 6 коронавирусов, способных заражать людей. Четыре из них (HCoV‐229E, HCoV‐OC43, HCoV‐NL63 и HKU1) вызывают симптомы, похожие на обычную простуду. Два — SARS‐CoV и MERS‐CoV могут заражать нижние дыхательные пути и с высокой вероятностью приводить к тяжелой пневмонии или даже смерти больного [11].
Атипичная пневмония и ближневосточный респираторный синдром
До начала 2000-х годов коронавирусы человека не воспринимались как серьезная опасность. В 2002 году в южных областях Китая произошла эпидемия тяжелого острого респираторного синдрома (SARS, также известного как атипичная пневмония), вызываемого новым на тот момент коронавирусом SARS-CoV [12]. Вспышка затронула более 8000 человек, чуть меньше 1000 из которых погибли [13]. В 2012 году распространение другого родственного вируса MERS-CoV привело ко вспышке заболевания ближневосточным респираторным синдромом, в результате которой заразилось 2066 человек и умерло 720. Эта эпидемия затронула преимущественно Саудовскую Аравию, хотя случаи были отмечены в 27 странах [14].
Оба этих коронавируса имели зоонозное происхождение — они передались людям, соответственно, от цивет (род мелких хищных млекопитающих) и верблюдов, которые, в свою очередь, получили эти вирусы от летучих мышей [8,15].
SARS-CoV-2 — возбудитель COVID-19
Как и заболевания SARS, COVID-19, вызываемый родственным вирусом SARS-CoV-2, опасен прежде всего поражением нижних дыхательных путей, в клетки которых вирус проникает, связываясь с тем же поверхностным рецептором ACE2.
Характеристики вируса
Многие детали о распространении SARS-CoV-2, его симптомах и эпидемиологических характеристиках, остаются предметами дискуссии и активного изучения. Вирус передается через капли крупного аэрозоля, который образуется при кашле или чихании. Однако вопрос, может ли вирус длительное время (часы) оставаться в воздухе в составе более мелких капель, остается открытым [19]. Важным путем передачи, по видимому, являются поверхности, на которых коронавирус может оставаться длительное время. Поэтому для снижения вероятности заражения важно часто мыть руки и меньше прикасаться к лицу. По всей видимости, распространять вирус могут даже те, у кого заражение проходит бессимптомно [19].
В Европейском центре по контролю за заболеваниями проанализировали распространение нового коронавируса в ходе вспышки в Италии. На основе полученных данных оценили базовое репродуктивное число для этого вируса (количество людей, которых заразит типичный заболевший). Это число оценивается в диапазоне от 2,76 до 3,25 [20]
Инкубационный период для вируса SARS-CoV-2 оценивается от 2 до 7 дней (для большинства случаев — 4 дня, хотя иногда доходит и до 14) [21]. Многие, если не большинство зараженных, болеют бессимптомно, хотя точно оценить процент бессимптомных случаев сложно. Из заболевших, у которых проявились симптомы, примерно 80% переносят болезнь сравнительно легко. Тяжелое состояние с гипоксией развивается примерно у 14%. Критическое состояние с дыхательной недостаточностью наблюдается примерно у 5% больных [19].
Летальность вируса, оценивается в диапазоне от 0,9 до 1,4 процентов, что приблизительно в 10 раз выше, чем у сезонного гриппа [22].
Происхождение
Последовательностью своего генома новый коронавирус SARS-CoV-2 больше всего похож на один из коронавирусов летучих мышей, который называется BatCoV RaTG13. Схожесть геномов составляет 96,2% [16]. Опираясь на это, ученые делают предположение об эволюционном родстве этих вирусов. Обнаружено также высокое (91,02%) сходство генома SARS-CoV-2 с геномом одного из коронавирусов панголинов Pangolin-CoV. Интересно, что несмотря на более высокое сходство нового коронавируса с вирусом летучих мышей, по строению ключевого поверхностного белка, отвечающего за связь и проникновение частицы в заражаемую клетку, SARS-CoV-2 больше похож как раз на вирус панголинов [17].
О строении вируса и процессе моделирования подробнее читайте в разделе о модели коронавируса SARS-CoV-2.
Молекулярный биолог Валерия Архипова и CGI художник Алексей Солодовников создали атомарную модель вируса SARS-CoV-2 и анимировали ее. По просьбе N + 1 они рассказывают, как много сил и времени заняла у них эта работа и какие данные им потребовались, чтобы построить изображение, которое одновременно было бы достоверным и при этом красивым.

Атомарная модель (каждый шарик — атом) SARS-CoV-2. Цветами обозначены: S- (бирюзовые), M- (зеленые) и E-белки (красные); мембрана (синий) и олигосахариды (оранжевые)
Наш вирус, помимо молекул оболочки (о том, почему они именно такие, подробнее расскажем ниже), собран из белков трех типов и олигосахаридных остатков на их поверхности.
S-белки (бирюзовые), они же спайк-белки или белки шипа — их вирус использует для того, чтобы прикрепляться к клеткам. Если эти белки свою работу делают хорошо, то вирус пробирается в клетку и переходит к размножению внутри нее.
Е-белки (красные), взаимодействуют с другими структурными белками, а также, будучи трансмембранными белками, образуют ионный канал, тем самым помогая сборке новых вирионов в комплексе Гольджи клетки-хозяина.
Другие модели
Изображения вируса, которые можно найти в открытом доступе, делятся на два типа: художественные иллюстрации без опоры на молекулярную биологию и работы ученых, где в приоритете — научная достоверность, а не эстетическая привлекательность.
Мы подумали, что можем совместить эти подходы: соблюсти достоверность и одновременно сделать изображение насыщенным. Показать и сложность, и красоту.
Конечно, работали мы не в информационном вакууме. Есть и другие модели — они нас вдохновляли и учили. Нам хотелось сделать не хуже или добавить что-то свое. Вот на какие модели мы обратили внимание, прежде чем заняться своей:
Это видео показало, что мы можем сделать нашу модель цветной, эстетически привлекательной. Выделить цветом разные части вируса, что улучшает восприятие изображения. Плюс здесь мы впервые увидели движение вируса — на изображениях, которые мы видели до этого, вирус всегда выглядел статично.
Данная работа показала, что мы можем уточнить структуру самого вируса, сделать ее атомарной. Это многократно повысило сложность модели, но и ее достоверность также возросла.
Тут мы увидели, что количество ключевых белков у вируса может быть очень различным. Мы приняли решение сосредоточиться на уточнении их количества, отдавая предпочтение самым новым источникам.
Эта работа начинается с 2D-иллюстрации ученого и художника Дэвида Гудселла (David S. Goodsell). Мы увидели, насколько важно придумать и выдержать цветовое решение модели вируса. В итоге, прежде чем мы дошли до финала, было отвергнуто 11 разных цветовых решений.
И последняя работа — это модель студии Visual Science. Эта модель показала, что наше решение сделать атомарную модель вируса — совершенно правильное. Также мы поняли, что число ключевых белков на поверхности вируса может сильно варьироваться. Мы приняли решение сосредоточиться на уточнении их количества, отдавая предпочтение самым новым научным источникам. Также мы увидели новую крупную задачу — сделать модель внутренней части вируса. Эту задачу мы оцениваем, как более сложную и не менее важную.
Все модели выше в большинстве своем сделаны крупными научными коллективами с привлечением значительных вычислительных ресурсов (облака, фермы, кластера). Нам было интересно, что может сделать наша команда из двух человек.
Источники данных
Часть белков на нашей иллюстрации взяты из базы Protein DataBase. Каждый белок — это отдельный файл, внутри которого хранятся координаты атомов белка, а также дополнительная информация: химический состав, аминокислотный, интенсивность колебаний атомов и тому подобное.
Для разных белков в Protein Database используют разные подходы к помещению атомов исследуемой молекулы в систему координат. Так, например, в прямоугольных декартовых координатах ось Z совмещается с максимальным значением момента инерции (для горизонтального расположения фрагмента белка) или совпадает с минимальным значением момента инерции (в случае вертикального расположения). Другой вариант используют для исследования длинных спиралевидных молекул: при этом ось X соответствует среднему значению момента инерции эллипсоида. Также широко используются сферические, цилиндрические и другие системы координат, которые потом пересчитываются в декартовы при создании PDB-файла.
По сути, формат PDB — это текстовый файл с данными про каждый атом. Вот как записывают, например, структуру Е-белка 5X29:

Фрагмент pdb-файла белка Е-белка 5X29. Например, для 878 атома координаты 0.035 -9.540 8.679 — это кислород
А вот как эта же информация выглядит уже в Houdini — программе, в которой данные из pdb-файлов преобразуются в 3D-модель, а затем в иллюстрацию.

Е-белок 5X29 в Houdini. Каждая точка — атом
PDB — очень ценный источник информации, без него у нас бы ничего не получилось. Это огромная — 178 938 биологических макромолекулярных структур на июнь 2021 года — международная база, которую пополняют ученые со всего мира, выкладывая данные своих исследований в открытый доступ.
Молекул оболочки нового коронавируса в полном виде в библиотеке PBD нет. Мы взяли оттуда липиды, из которых она может состоять, и построили ее самостоятельно. Вирус собирает свою оболочку из фрагментов мембраны клетки-хозяина — этот суперкапсид способствует повышению заразности вируса. Суперкапсид нашего вируса построен из липидов мембраны клетки эпителия.
Далее нам нужно было определить количество объектов, из которых состоит вирус, и предсказать их движение. В нашей модели мы отобразили все атомы, которые есть в исходных данных, без упрощения. Вот что у нас получилось сначала:

Промежуточная модель коронавируса SARS-COV-2
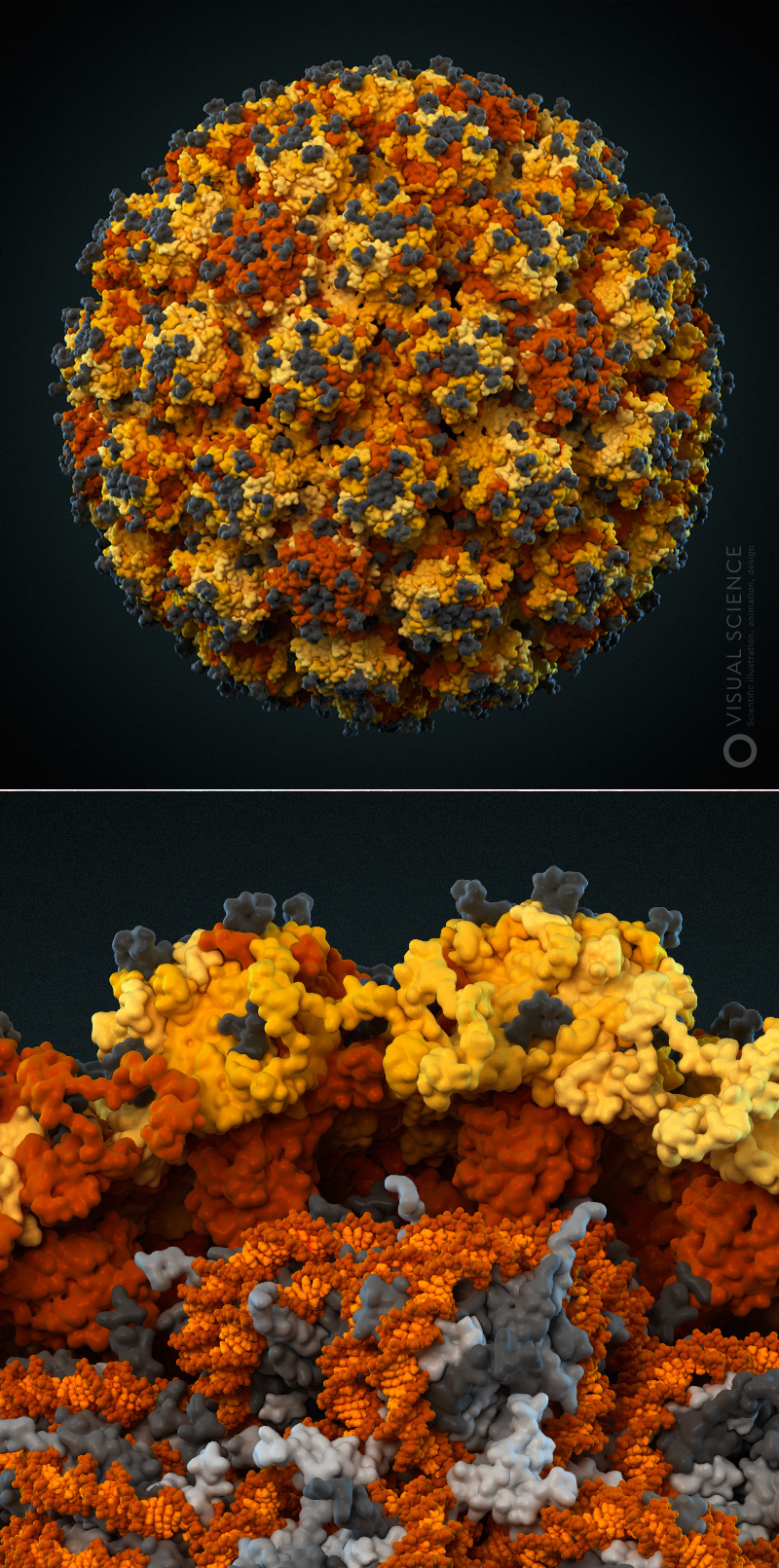
Затем нам удалось получить консультацию экспертов. Это позволило повысить точность нашей модели. Мы чрезвычайно признательны Николаю Никитину с кафедры вирусологии МГУ и Софье Борисевич из лаборатории химической физики Уфимского Института химии РАН. Вместе с ними мы уточнили количество S-белков, которое зависит от размера вириона (сам он составляет 50-200 нанометров). По последним данным их не 90, как было в первой версии нашей иллюстрации, а меньше — от 26 до 41-го. На нашей модели S-белков 38.
Также большим сюрпризом для нас оказалась подвижность S-белка:

Иллюстрация, показывающая сгибы и вращение S-белка
Choi et al. / Journal of Chemical Theory and Computation, 2021
Помимо этого, мы выяснили, что M-белки расположены на оболочке намного плотнее — их стало около 1000 штук на вирион. А E-белков, как минорной компоненты, напротив, очень мало. В нашем случае их 15 штук на вирион.
Как моделировали
Вот как выглядит 3D-модель вируса в Houdini:

Houdini. Собранная 3D модель. Геометрическое представление. Без материалов и света
Houdini используется для создания спецэффектов в кино и рекламе: взрывы, дым, вода, огонь, разрушения. Подошел он и для наших задач. Внешняя часть вируса нашей модели — приблизительно 25 000 000 атомов. Просчет изображения с материалами и светом выполнялся на GPU рендере RedShift. По сравнению с CPU (16-ядерный AMD против 3080 NVIDIA), прирост по скорости около семи раз. Модель вируса может быть посчитана в очень больших разрешениях. Рендер для фрагмента, на котором хорошо видны атомы, сделан в 8000х8000 пикселей, что эквивалентно 1300х1300 миллиметрам при печати в 150 точек на дюйм.
Вирус рендерился в черно-белом виде. И дальше уже по маскам красился вручную в программе для композитинга и монтажа — DaVinci Resolve. Такой подход позволил подобрать цветовую гамму без пересчета модели. Всего было испробовано порядка десяти вариантов цветовых решений. Ниже — не раскрашенная модель вируса.


Наша модель SARS-CoV-2. Фрагмент из полного изображения 8000х8000 пикселей (130х130 сантиметров). Хорошо видны атомы.
Итого
Моделированием внутренней структуры вируса мы не занимались. Это около 30 000 нуклеотидов РНК, N-белков и других элементов. Эта работа сложнее, чем моделирование внешнего вида вируса. Во многом это связано с отсутствием моделей белков и тем, что для этого нужно решить нетривиальную задачу укладки нити РНК в полости сферы вируса.
Повысить точность нашей модели можно с привлечением молекулярной динамики. Тогда оболочку можно будет изобразить точнее, как и само движение атомов. Молекулярная динамика позволяет получить очень точное представление о движении и взаимодействии частей вируса как друг с другом, так и с внешней средой. Но МД требует специальных навыков работы и очень требовательна к вычислительным ресурсам. У нас нет необходимых мощностей, чтобы работа была выполнена за приемлемые сроки. Поэтому движения атомов и частей вируса показаны условно.
В открытых источниках мы не нашли информации о структуре М-белка. Поэтому и он тоже показан условно. Размеры атомов усреднены.
Вся работа заняла у нас приблизительно полгода: она велась параллельно с другими проектами, после основной работы, домашних дел, с перерывами на болезни и праздники и поправкой на разные часовые пояса.
Мы получили модель вируса, изображения с ним и анимацию его условного движения. Что дальше, что с этим делать и какая от этого польза?
Во-первых, это красиво. Во-вторых, мы показываем сложность объектов на микроскопическом, атомарном уровне. Наша работа позволяет эту сложность увидеть. И последнее — пожалуй, самое важное. Наш проект показывает, что и один человек может сделать очень многое. А значит, может каждый из нас. Дерзайте!
Список литературы
1. Surya, W., Li, Y., Torres, J. Structural model of the SARS coronavirus E channel in LMPG micelles. // Biochim. Biophys. Acta - Biomembr. – 2018. – Vol. 1860. – N. 6. – P. 1309–1317.
2. Koppisetti, R. K., Fulcher, Y. G., Jurkevich, A., Prior, S. H., Xu, J., Lenoir, M., Overduin, M., Van Doren, S. R. Ambidextrous binding of cell and membrane bilayers by soluble matrix metalloproteinase-12. // Nat. Commun. – 2014. – Vol. 5. – P. 1–14.
3. Hillen, H. S., Kokic, G., Farnung, L., Dienemann, C., Tegunov, D., Cramer, P. Structure of replicating SARS-CoV-2 polymerase. // Nature. – 2020. – Vol. 584. – N. 7819. – P. 154–156.
4. Harris, L. J., Larson, S. B., Hasel, K. W., McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. // Biochemistry. – 1997. – Vol. 36. – N. 7. – P. 1581–1597.
5. Noreng, S., Bharadwaj, A., Posert, R., Yoshioka, C., Baconguis, I. Structure of the human epithelial sodium channel by cryo-electron microscopy. // Elife. – 2018. – Vol. 7. – P. 1–23.
6. Almond, A., DeAngelis, P. L., Blundell, C. D. Hyaluronan: The Local Solution Conformation Determined by NMR and Computer Modeling is Close to a Contracted Left-handed 4-Fold Helix. // J. Mol. Biol. – 2006. – Vol. 358. – N. 5. – P. 1256–1269.
7. Hurdiss, D. L., Drulyte, I., Lang, Y., Shamorkina, T. M., Pronker, M. F., van Kuppeveld, F. J. M., Snijder, J., de Groot, R. J. Cryo-EM structure of coronavirus-HKU1 haemagglutinin esterase reveals architectural changes arising from prolonged circulation in humans. // Nat. Commun. – 2020. – Vol. 11. – N. 1. – P. 1–10.
8. Yan, Renhong, Yuanyuan Zhang, Yaning Li, Lu Xia, Yingying Guo, Q. Z. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. // Science (80-. ). – 2020. – Vol. 3. – N. 3. – P. 1–8.
9. Javitt, G., Khmelnitsky, L., Albert, L., Bigman, L. S., Elad, N., Morgenstern, D., Ilani, T., Levy, Y., Diskin, R., Fass, D. Assembly Mechanism of Mucin and von Willebrand Factor Polymers. // Cell. – 2020. – Vol. 183. – N. 3. – P. 717-729.e16.
10. Daniel Wrapp, Nianshuang Wang, Kizzmekia S. Corbett, Jory A. Goldsmith, Ching-Lin Hsieh, Olubukola Abiona, B. S. G., McLellan, and J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. // Science (80-. ). – 2020. – Vol. 21. – N. 1. – P. 1–9.
11. Wang, M. Y., Zhao, R., Gao, L. J., Gao, X. F., Wang, D. P., Cao, J. M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. // Front. Cell. Infect. Microbiol. – 2020. – Vol. 10. – N. November. – P. 1–17.
12. Yao, H., Song, Y., Chen, Y., Wu, N., Xu, J., Sun, C., Zhang, J., Weng, T., Zhang, Z., Wu, Z., Cheng, L., Shi, D., Lu, X., Lei, J., Crispin, M., Shi, Y., Li, L., Li, S. Molecular Architecture of the SARS-CoV-2 Virus. // Cell. – 2020. – Vol. 183. – N. 3. – P. 730-738.e13.
13. Oostra, M., de Haan, C. A. M., de Groot, R. J., Rottier, P. J. M. Glycosylation of the Severe Acute Respiratory Syndrome Coronavirus Triple-Spanning Membrane Proteins 3a and M. // J. Virol. – 2006. – Vol. 80. – N. 5. – P. 2326–2336.
14. B.W. Neuman, M. J. B. Supramolecular Architecture of the Coronavirus Particle. // Adv. Virus Res. – 2020. – Vol. 96. – P. 1–27.
15. Neuman, B. W., Kiss, G., Kunding, A. H., Bhella, D., Baksh, M. F., Connelly, S., Droese, B., Klaus, J. P., Makino, S., Sawicki, S. G., Siddell, S. G., Stamou, D. G., Wilson, I. A., Kuhn, P., Buchmeier, M. J. A structural analysis of M protein in coronavirus assembly and morphology. // J. Struct. Biol. – 2011. – Vol. 174. – N. 1. – P. 11–22.
16. Yu, A., Pak, A. J., He, P., Monje-Galvan, V., Casalino, L., Gaieb, Z., Dommer, A. C., Amaro, R. E., Voth, G. A. A multiscale coarse-grained model of the SARS-CoV-2 virion. // Biophys. J. – 2021. – Vol. 120. – N. 6. – P. 1097–1104.
17. Yao, H., Song, Y., Chen, Y., Wu, N., Xu, J., Sun, C., Zhang, J., Weng, T., Zhang, Z., Wu, Z., Cheng, L., Shi, D., Lu, X., Lei, J., Crispin, M., Shi, Y., Li, L., Li, S. Molecular architecture of the SARS-CoV-2 virus. // Cell. – 2020. – Vol. 183. – N. 3. – P. 730–738.
18. Choi, Y. K., Cao, Y., Frank, M., Woo, H., Park, S. J., Yeom, M. S., Croll, T. I., Seok, C., Im, W. Structure, Dynamics, Receptor Binding, and Antibody Binding of the Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein in a Viral Membrane. // J. Chem. Theory Comput. – 2021. – Vol. 17. – N. 4. – P. 2479–2487.

Под катом первая часть рассказа о нашем опыте создания научно достверных моделей вирусов.
Мир молекулярных машин и вирусов предлагает массу интересных вызовов CG командам. Проблема в том, что пока не существует универсальной научной методики, которая позволила бы полностью описать строение вирусной частицы. Для того, чтобы описать устройство вируса нужно пользоваться множеством методов, которые дают представление об отдельных кусках финального паззла. Электронная микроскопия позволяет оценить размеры и очертания вирионов, рентгеноструктурный анализ способен описать отдельные белки или их фрагменты, а молекулярно-биологические и биохимические методы дают сведения о том, сколько каких молекул входит в состав вируса и как они между собой взаимодействуют. При этом создается несколько парадоксальная ситуация: многие вирусы изучены очень подробно и в деталях, но не существует изображений, которые давали бы научно достоверное и полное представление о том, как они устроены.
Например, современные электронные микрофотографии вирусных частиц гриппа выглядят так (источник).
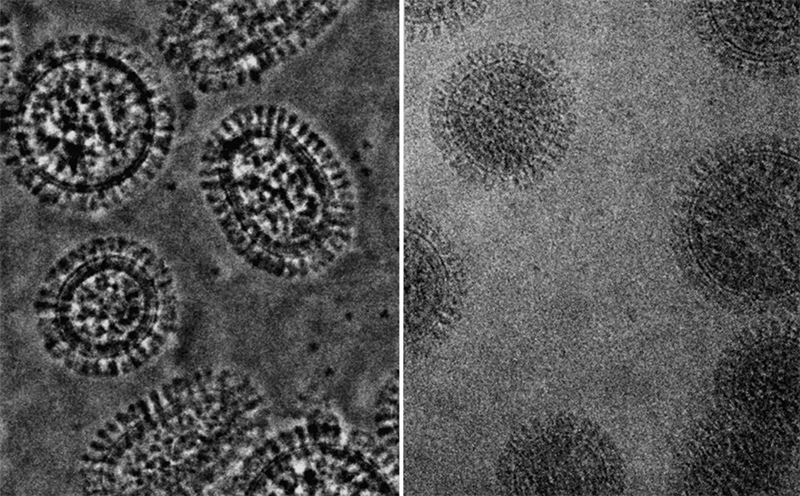
Визуализация данных криоэлектронной микроскопии геномного комплекса вируса гриппа А и реконструкция упаковки РНК (желтая лента) белками В и С. Работу с этими данными опубликовала в конце 2012 года в журнале Science группа вирусологов из Мадрида, которые помогли нам в создании модели вируса гриппа A/H1N1.
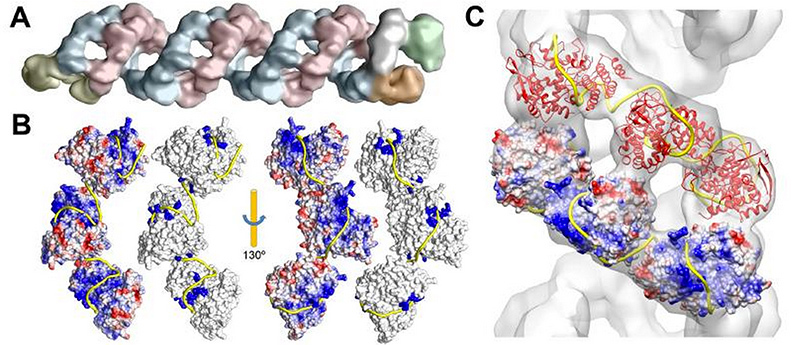
Собрать всю доступную информацию технически возможно. Но ее систематизация, обработка и перевод в 3Д модель требует командного подхода. При этом даже компетентный научный консультант не может обладать полным багажом узкоспециальных знаний по теме, поэтому к проекту важно подключить ученых, посвятивших работе с тем или иным вирусом всю свою карьеру. Моделлер без биологического образования не разберется в опубликованных научных данных и структурах белков из Protein Data Bank, а также не сможет корректно достроить модели молекул при помощи молекулярной динамики, где это необходимо (приблизительно 80-90% белков, с которыми мы сталкиваемся, имеют неполное описание пространственной струкутры на 10-90%). Ученый же, даже обладая всеми сведениями в отдельности, не может собрать и визуализировать полную модель в профессиональных пограммах для трехмерного моделирования. По нашему опыту, только тесное взаимодействие этих специалистов может дать аккуратный и информативный результат.
Изображение вируса гриппа с детализацией до атомов. Все белки и белковые комплексы в составе частицы, а также их количественные соотношения и положение соответствуют опубликованным в научной литературе данным (подписи всех компонентов). Модель создана при участии Хайме Мартин-Бенито и коллег (Испанский национальный центр биотехнологий, Мадрид, Испания). 2013 год.
Внутреннее устройство вируса иммунодефицита человека. Виден край мембранной оболочки, белки, присутствующие внутри вириона, капсид и фрагменты РНК вируса, в нем заключенные (подписи всех компонентов). Модель создана при участии Егора Воронина (Global HIV Vaccine Enterprise). Приз за лучшую научную иллюстрацию на конкурсе Science and Engineering Visualization Challenge в 2011 году.

Модель предполагаемой укладки генома вируса папилломы человека. Модель создана при участии Кристофера Бака (Национальный институт рака, США). 2012 год.
Частица и отдельные белки вируса Эбола. Модель создана при участии Рональда Харти (Университет Пеннисльвании, США). Honorable mention конкурса Science and Engineering Visualization Challenge в 2010 году. Экспозиция салона Ассоциации медицинских иллюстраторов в Торонто в 2012 году.
Наша студия несколько лет назад запустила некоммерческий проект, суть которого в моделировании и визуализации наиболее распространенных и опасных вирусов человека. Мы назвали его Viral Park, или “Зоопарк вирусов”. Проект пока включает четыре вирусные модели, еще несколько находятся в разработке, а в планах сделать серию из примерно двадцати вирионов. За время работы над проектом мы успели освоить и наладить процесс, выделив в нем ряд этапов:
- Обзор литературы и систематизация обнаруженных данных
- Молекулярное моделирование и динамика
- Сборка полной модели из отдельных элементов
- 3D визуализация и дизайн
- Создание материалов на основе модели от плакатов до приложений, виджетов и пластиковых моделей.
В этом посте мы немного расскажем о первом этапе нашей работы.
Сбор информации об изучаемой теме — это задача, которую ученые решают постоянно. Невозможно сделать новый проект, не зная того, что опубликовали до тебя. Для этого надо найти и проанализировать сначала обзорные, а потом и исследовательские публикации по интересующему вопросу. Та же схема работает, когда собирается информация о строении вирусов. Благодаря базам естественнонаучных публикаций основных мировых журналов PubMed и Google Scholar этот процесс можно организовать весьма эффективно. Если нужна вводная информация о биологии вируса, можно воспользоваться сайтом Viral Zone а много данных по отдельным белкам доступно в базе данных Uniprot. Структуры белков или их фрагментов, полученные разными коллективами ученых при помощи методов ядерного магнитного резонанса и рентгеноструктурного анализа, доступны в уже упомянутом Protein Data Bank в виде координат всех атомов или, в ряде случаев, только альфа-атомов цепочки белка.
Задачей для ученого в процессе создания модели вируса являются сбор, обработка и подготовка всей информации в том виде, который будет удобен для работы остальных членов команды. Нужно составить полный список всех типов молекул, которые образуют частицу, и всех их взаимодействий. Помимо белков это могут быть липиды мембраны и молекулы вирусного генома, представленные ДНК или РНК. Дальше надо понять, в каких количествах молекулы представлены в частице, и какие места они занимают. Эта наиболее сложная для поиска и часто противоречивая и неполная информация, поскольку разные методы могут давать разные оценки. Для уточнения тех или иных вопросов мы связываемся с авторами статей, в которых они обсуждаются. Это вполне принятая практика в научном сообществе, и ученые часто с удовольствием, а иногда без идут на контакт и порой делятся своими гипотезами и даже неопубликованными данными, как это было при работе над моделью Гриппа в случае с уже упомянутыми испанскими вирусологами.
Результатом исследования литературы должна стать максимально подробная вербальная картина будущей модели. Надо понимать что, в каких количествах и каким образом упаковано в вирусной частице. Это можно свести в описание, таблицу количеств и взаимодействий и план модели в нужном масштабе.
Дальнейшие этапы работы подразумевают получение трехмерных моделей всех нужных компонентов. Одной из проблем тут является то, что не для всех белков и их комплексов могут быть доступны атомные структуры. Существенную часть вирусных белков ученым просто еще не удалось описать. В нашей работе мы используем методы структурной биоинформатики, чтобы заполнить этот пробел. Об этом мы расскажем в следующих постах. Также постараемся раскрыть детали того, как происходит сборка полной модели, ее визуализация и создание образовательных пособий и виджетов на основе полученного результата.
Мы считаем, что у такого детального подхода к моделированию молекулярно-биологических объектов большие перспективы с точки зрения его применения в образовании, популяризации науки и научной коммуникации. В пользу этого говорит и то, что такие модели получают высокие оценки на крупных международных конкурсах научной иллюстрации и дизайна, положительные отзывы известных коллег, а включить такие изображения в свои презентации бывает приятно даже Франсуазе Барре-Синусси, получившей Нобелевскую премию за открытие ВИЧ.
В продолжении темы, помимо моделирования вирусов в рамках Зоопарка вирусов, мы обсудим сферу научной и медицинской иллюстрации в целом, поговорим о том, почему это актуально, чем это отличается от набирающего популярность научного исскуства, или Science Art, и как это поможет сделать мир лучше а науку понятнее.
Коронавирус типа 2019-nCoV, после вспышки заболевания в китайском городе Ухань, стремительно распространяется по миру. На момент написания оригинальной статьи (30 января 2020 года) сообщалось о более чем 9000 заражённых и о 213 умерших, на сегодня (10 февраля 2020 года) сообщается уже о 40570 зараженных, 910 человек умерло. Случаи заражения коронавирусом выявлены во Франции, в Австралии, в России, в Японии, в Сингапуре, в Малайзии, в Германии, в Италии, в Шри-Ланке, в Камбодже, в Непале и во многих других странах. Никто не знает о том, когда вирус будет остановлен. Пока же число подтверждённых случаев коронавируса лишь растёт.
Автор статьи, перевод которой мы сегодня публикуем, хочет рассказать о том, как, с использованием Python, создать простое приложение для отслеживания распространения коронавируса. После завершения работы над этим приложением в распоряжении читателя окажется HTML-страница, которая выводит карту распространения вируса и ползунок, который позволяет выбирать дату, по состоянию на которую данные выводятся на карту.
Интерактивная карта распространения коронавируса типа 2019-nCoV
Здесь будут использованы такие технологии, как Python 3.7, Pandas, Plotly 4.1.0 и Jupyter Notebook.
Импорт библиотек
Начнём работу с импорта в проект, основанный на Jupyter Notebook, библиотек Plotly и Pandas.
Обработка данных
Данные, которыми мы тут пользуемся, можно найти здесь. Это — расшаренная таблица из документов Google, которая ежедневно обновляется. Огромное спасибо всем тем, кто поддерживает её в актуальном состоянии! Вы делаете очень нужную работу.
Мы будем читать данные, используя метод Pandas read_csv . Но прежде чем загружать данные из таблицы, воспользовавшись ссылкой на неё, нам нужно поработать с этой ссылкой. Сейчас она выглядит так:
Нам нужно заменить выделенный фрагмент ссылки, приведя ссылку к такому виду:
В следующем коде мы инициализируем переменную url , записывая в неё ссылку на данные, читаем данные с использованием метода read_csv и записываем в пустые ячейки, содержащие NaN , значения 0 .
Понимание того, как устроены структуры данных, которые мы используем, чрезвычайно важно на данном шаге работы, так как это определяет то, какой подход к обработке данных мы применим. Просмотрим данные, воспользовавшись командой data.head() . Это приведёт к выводу первых 5 строк таблицы.

Первые 5 строк данных по коронавирусу
В левом нижнем углу можно видеть сведения о том, что в таблице данных имеется 47 столбцов. Вот имена первых пяти столбцов: country , location_id , location , latitude и longitude . Другие столбцы представляют собой пары, имена которых построены по следующей схеме: confirmedcase_dd-mm-yyyy и deaths_dd-mm-yyyy . Общее число столбцов в таблице на момент написания этого материала было 47. Это означает, что в моём распоряжении были данные за 21 день ((47-5)/2=21). Если начальной датой сбора данных было 10.01.2020, то конечной датой было 30.01.2020.
Имена первых пяти столбцов таблицы не меняются, но с течением времени в таблицу будут добавляться колонки с новыми именами. То, что будет выводить наша интерактивная карта, представляет собой визуализацию распространения коронавируса с возможностью указания дня, по данным которого формируется карта. Поэтому нам нужно разделить весь набор данных, выбрав из него сведения по каждому дню и учитывая то, что первые 5 столбцов таблицы не меняются, и то, что каждый день описывается двумя столбцами. Затем, если внимательнее присмотреться к данным, например — к данным за 10.01.2020, то окажется, что этой дате соответствует множество строк. На самом деле, на эту дату обнаружение коронавируса было подтверждено лишь в одном месте, что отмечено соответствующим числом. Во всех других строках на эту дату содержатся лишь нули. Это означает, что нам нужно исключить эти строки из процесса построения карты.
Процесс подготовки данных к визуализации производится в цикле.
В процессе работы выходные данные каждого набора данных добавляются к Scattergeo-графику с использованием fig.add_trace . На момент написания материала данные, на основе которых будут строиться изображения, представлены 21 объектом. Проверить это можно, воспользовавшись командой fig.data .
Создание слайдера
Здесь мы создадим слайдер, с помощью которого организован выбор данных, визуализируемых на карте. Вот соответствующий код:
Код слайдера состоит из двух основных фрагментов. Первый — это цикл, в котором заполняется список steps , используемый при перемещении бегунка слайдера. При перемещении слайдера выполняется визуализация соответствующего набора данных и скрытие того, что было выведено до этого. Вторая часть кода — это включение сконструированного ранее списка steps в объект слайдера. Когда слайдер перемещается — осуществляется выбор соответствующего элемента из steps .
Вывод карты и сохранение её в виде HTML-файла
Сейчас мы подошли к финальной части материала. Тут мы поговорим о том, как выводить карту, и о том, как сохранять её в формате HTML. Вот код, реализующий эти операции:
Когда карта выводится на экран, видимым оказывается визуальное представление первого набора данных. Затем мы делаем так, чтобы содержимое карты обновлялось бы в соответствии с положением слайдера. Тут же мы задаём заголовок карты и настраиваем её высоту. На последнем шаге мы выводим карту, пользуясь методом fig.show , после чего сохраняем её в HTML с помощью метода go_offline.plot .
Полный код проекта
Вот полный код проекта, позволяющего создать карту распространения коронавируса типа 2019-nCoV. Обратите внимание на то, что последнюю строчку, ответственную за сохранение HTML-варианта карты, нужно отредактировать, заменив указанный там путь на тот, который актуален для вас.
Итоги
Мы завершили разбор руководства, посвящённого созданию интерактивной карты визуализации распространения коронавируса типа 2019-nCoV. Проработав этот материал, вы узнали о том, как читать данные из общедоступных таблиц Google, как выполнять обработку данных с помощью Pandas, и как визуализировать эти данные на интерактивной карте с использованием слайдера и Plotly. Результат работы проекта в виде HTML-страницы можно загрузить отсюда. Информация, выводимая на карте, зависит от таблицы с данными. Каждый раз, когда выполняется код проекта, карта обновляется, на ней становятся доступными свежие данные из таблицы. Это — очень простая карта. Существует множество путей её улучшения. Например, её можно дополнить дополнительными графиками, некими сводными данными и так далее. Если вам это интересно — вы вполне можете сделать всё это и многое другое самостоятельно.
Реалии жизни накладывают свой отпечаток и на творчество. Едва ли пару месяцев тому назад вы думали о том, что будете искать бесплатную схему вязания коронавируса крючком, но сегодня этот запрос уже никого не удивит. Кому-то хочется сделать познавательную игрушку для ребенка, кому-то нужны наглядные пособия для уроков в школе или детском саду, одним хочется обзавестись памятным брелком, другие считают эту идею неплохим сувениром в подарок любимому доктору, любимой клинике, любимой медсестре. Какие бы причины не руководили вами, обратите внимание на предлагаемую подборку схем вязания коронавируса крючком - это и забавные, и серьезные, и веселые, и реалистичные, и прикольные, и вполне настоящие модели клетки, которую сейчас так многие боятся. Работаем?

Как связать коронавирус своими руками - 5 бесплатных схем:
1. Коронавирус реалистичный

Если вы ищете, как сделать клетку коронавируса своими руками для презентаций и учебных лекций, этот вариант - несомненно один из лучших. Во-первых, схема довольно простая - вы не будете сидеть над ней часами, разбираясь, как связать тот или иной элемент, а во-вторых, результат действительно очень и очень похож на первоисточник: он дает представление о том, как на самом деле выглядит клетка этого нашумевшего вируса.
2. Коронавирус забавный

Не считаете, что клетка коронавируса может быть забавной? Слушайте, но, конечно же, тема серьезная, и шутки тут не особо и уместны, однако, если вы не склонны драматизировать происходящее и умеете находить то-то хорошее даже в плохом, обратите все-таки внимание на этот проект. Он несложный, доступный, интересный и - да! - результат вызывает улыбку, и порою это не так и плохо, даже если кажется неуместным.
3. Коронавирус глазастый

Между прочим, многие психологи утверждают, что людям, которые сейчас панически боятся заразиться коронавирусом и чувствуют повышенный уровень тревоги, необходимо порою просто посмотреть своим страхам в лицо. Вероятно, такая техника подойдет не всем, но попробовать-то можно, верно? Попробуйте связать клетку коронавируса своими руками и заглянуть ей в глаза - что она сказала бы вам, если бы могла?
4. Коронавирус-злюка

Хорошая игрушка для детей, если вы хотите объяснить им, что такое коронавирус, чем страшен, почему его так опасаются миллионы людей на планете и каковы основные правила поведения и гигиены существуют для того, чтобы минимизировать риски заражения. Да, коронавирус может привести к очень и очень неприятным последствиям, он злюка и вообще не очень приятный тип, однако, соблюдая правила, можно избежать с ним встречи.
5. Коронавирус сердитый

Вязание - это стрессотерапия. Если вы чувствуете, что нужно сбросить напряжение, расслабиться, позволить себе отдых и переключиться на что-то другое, попробуйте предложить самой себе повязать крючком. Отзывается? Тогда почему бы не связать клетку коронавируса? Проработать страхи, создать для детей наглядный образец или просто увековечить свои воспоминания об этом периоде жизни таким вот нестандартным способом?
Читайте также: