
Признаки активации апоптоза на фоне вирусной инфекции что это
Обновлено: 19.04.2024
Численность популяции клеток в организме связана с двумя противоположно направленными процессами: митотическим размножением и гибелью клеток. Длительное время значение гибели клеток недооценивалось, однако в последнее время интерес к механизмам ее реализации значительно повысился, и эта проблема стала одной из наиболее интенсивно изучаемых областей биологии. На сегодняшний день установлено, что нарушение контроля клеточной гибели ведет к сдвигам гомеостаза и развитию различных патологических состояний.
На клеточном уровне постоянно протекающие деление и рост должны сопровождаться альтернативным процессом удаления старых, поврежденных, мутировавших и других нежелательных для организма клеток. Высокорегулируемую форму программированной смерти клетки с характерными морфологическими и биохимическими признаками определяют как апоптоз (греческое слово, соответствующее русскому "листопаду": аро - отделение, ptosis - падение).
Апоптозу принадлежит важнейшая роль как в физиологических, так и в патологических условиях, ввиду того, что и подавление, и неадекватное усиление апоптоза ведет к патологическим изменениям органов и тканей. В то время как избыточная активация апоптоза, наблюдаемая, в частности, при инфицировании клеток печени гепатотропными вирусами, обусловливает разрушение печеночной ткани, ослабление апоптотической гибели клеток (вызванное, к примеру, мутацией гена, кодирующего проапоптогенный белок р53) служит одним из важнейших факторов канцерогенеза.
Что касается гепатита, наиболее важным диагностическим и прогностическим признаком при многих его формах считается некроз паренхимы, который, однако, значительно отличается от типичных признаков омертвения, наблюдаемых морфологом в других органах. Прежде всего в ткани печени отсутствуют некротизированные гепатоциты, а видны лишь участки печеночной дольки, замещенные мононуклеарными инфильтратами. Вторая особенность - отсутствие полиморфноядерных лейкоцитов (за исключением острого алкогольного гепатита) -стереотипной реакции на некроз во всех тканях. Следовательно, в большинстве случаев хронического поражения печени, в том числе при инфекции гепатотропными вирусами, основным механизмом гибели клеток служит апоптоз. За много лет до открытия апоптоза был описан характерный гистологический признак вирусного гепатита - округлые гомогенные эозинофильные образования, часто содержащие пикнотичное ядро. Эти образования, названные тельцами Каунсильмена, представляют собой не что иное, как гепатоциты в состоянии апоптоза [2]. Кардинальные отличия апоптоза от некроза приведены в таблице 1.
Таблица 1. Основные характеристики апоптоза и некроза
Апоптоз
Некроз
Физиологический или патологический
Нерегулируемый или
слабо регулируемый
Плазматическая мембрана
интактна до поздней стадии
Плазматическая мембрана
разрушается в начальной стадии
Инфильтрация полиморфноядерными
лейкоцитами отсутствует
или минимальна
Лейкоцитарная инфильтрация
всегда присутствует
Кариопикноз, кариорексис,
фрагментация ДНК
Набухание (онкоз) цитоплазмы
и митохондрий
Образование апоптозных телец
с последующим их фагоцитозом
или вторичным некрозом
Разрушение
и дезинтеграция клетки
Первая стадия апоптоза - лиганд-рецепторное взаимодействие. Представление о функционировании рецепторов клеточной гибели служит теоретической базой для разработки оптимальной стратегии патогенетического лечения различных заболеваний, в том числе вирусных гепатитов.
Рецепторы клеточной гибели включают Fas-peцептор (Fas-R), TNF-R1, TNF-R2, "рецептор смерти-3" (DR-3) и 4 так называемых TNF-ассоциированных апоптоз-индуцирующих лиганд-рецептора [5]. Наиболее хорошо среди них изучены Fas-R и TNF-R1.
Fas-R (APO-1/CD95) экспрессируется в печени на гепатоцитах, холангиоцитах, активированных звездчатых ретикулоэндотелиоцитах и клетках Купфера и существует в мембраноассоциированной и растворимой формах. Растворимая форма Fas-R, вероятно, служит для связывания Fas-лиганда (Fas-L) не только на СD8+-цитотоксических лимфоцитах и NK-клетках, но и СD4+-Т-лимфоцитах-хелперах 1-го типа, которые, как было недавно установлено, также могут проявлять цитотоксические свойства [I]. Связывание Fas-L растворимым Fas-R уменьшает повреждение печени цитотоксическими иммунными клетками. Это подтверждается экспериментом S. Krams по инъекции мышам антител к Fas-R, что приводило к смерти животных от фульминантной печеночной недостаточности [8].
Экспрессия Fas-R на мембране гепатоцитов индуцируется рядом провоспалительных цитокинов, таких как интерлейкины (ИЛ-1, -2, -6), интерфероны (ИФН-γ), факторы некроза опухоли (TNF-α) и др. Таким образом, вероятно, что воспаление любой природы может способствовать Fas-R-зависимому повреждению печени. Более того, цитокины стимулируют увеличение количества молекул Fas-L на Т- и NK-лимфоцитах. Интересные данные были получены в работе, в которой культура.крысиных гепатоцитов подвергалась воздействию активированных ИЛ-2 NK-клеток и экспериментальных ингибиторов каспаз. Подавление активности каспаз предотвращало апоптоз гепатоцитов, но при этом усиливалась их гибель посредством некроза, что указывает на определенный баланс между этими путями элиминации клеток, осуществляемыми иммунной системой [5].
Связывание Fas-R ведет к его олигомеризации и активации адапторного белка FADD, что в свою очередь вызывает активацию специфического протеолитического фермента каспазы 8; этот ступенчатый процесс представляет собой основной механизм инициации как физиологического,так и патологического апоптоза клеток печени. На этом этапе дальнейшее развитие апоптоза может быть заблокировано активацией ряда факторов, таких как I-FLICE (эндогенная доминантно-негативная форма каспазы 8), bcl-2 и т.н. Х-связанные ингибиторы апоптоза [1, 2, 5].
Внутриклеточный домен TNF-R1 также интенсивно экспрессируется на гепатоцитах и клетках Купфера. Его экспрессия резко повышается при гепатите любой этиологии (вирусный, алкогольный, аутоиммунный и др.). Исследования F. Su и соавторов [10] продемонстрировали, что НВх-протеин вируса гепатита В сенсибилизирует культуру гепатоцитов к TNF-α-индуцированной цитотоксичности. Персистенция HBsAg также сенсибилизирует гепатоцит к TNF-опосредованному апоптозу.
Апоптоз, индуцированный связыванием TNF-α с TNF-R1, подобно взаимодействию Fas-R - Fas-L, требует олигомеризации рецептора и может осуществляться через путь FADD - каспаза 8, а также сходный с ним белок TRADD. Интересно, что гиперэкспрессия TRADD ведет не только к запуску процессов апоптоза, но и к активации ядерного фактора кВ (NFKB), который предотвращает TNF-индуцированную гибель клетки [2, 5].
Следует заметить, что даже в здоровой печени NK- и CD8+-T-лимфoциты печеночных синусоидов содержат больше мРНК ИЛ-15, -18, TNF-α, ИФН-γ по сравнению с периферическими клетками, что свидетельствует о повышенной готовности к осуществлению апоптоза гепатоцитов в случае возникновения их изменений, например, при инфицировании вирусом [7].
Экспрессия Fas-R существенно повышена на мембране гепатоцитов, инфицированных вирусами В и С, и тесно коррелирует с их гистологической активностью [б]. При вирусном гепатите апоптоз может быть как результатом прямого воздействия вируса, так и опосредованным иммунной реакцией. Запуск процессов апоптоза при проникновении в гепатоцит вируса следует рассматривать как своего рода защитный механизм, так как в мертвой клетке репликация вируса становится невозможной. Поэтому "в интересах" вируса - подавить апоптоз и сохранить клетки жизнеспособными. И действительно, некоторые кодируемые вирусами белки обладают антиапоптозной активностью, которая осуществляется подавлением функции проапоптогенного белка р53, инактивацией каспаз, а также усиленной экспрессией мощного ингибитора апоптоза bcl-2. Интересно, что в норме bcl-2 обнаружен в печени только в эпителии желчных протоков, постоянно контактирующих с желчью, но не в гепатоцитах [2].
Однако чаще причиной апоптоза при инфекции гепатотропными вирусами служит не прямое цитотоксическое действие вируса, а иммунная реакция NK- и Т-лимфоцитов на его антигены, расположенные на поверхности инфицированных гепатоцитов.
Т-лимфоциты могут вызывать апоптоз в клетках печени двумя путями. Первый реализуется за счет выброса из Т-клеток перфорина, который образует поры в плазматических мембранах гепатоцитов. Через них в клетки проникают гранзимы - протеазы Т-лимфоцитарных гранул, вызывающие расщепление ряда внутриклеточных ферментов, в том числе каспаз, запускающих апоптозный каскад.
Второй путь осуществления апоптоза под воздействием активированных Т-лимфоцитов связан с рассматривавшимся выше взаимодействием Fas-R -Fas-L (см. рисунок). Как уже упоминалось, Fas-R усиленно экспрессируется на мембране инфицированных гепатотропными вирусами гепатоцитов, но особенно часто их обнаруживают на гепатоцитах, окруженных лимфоцитами на границе ступенчатого некроза и паренхимы [6]. В свою очередь, на соответствующих лимфоцитах выявляется повышенная экспрессия Fas-L. О связи экспрессии Fas и последующего апоптоза с действием вируса гепатита С свидетельствует и то, что после успешного лечения а-интерфероном количество Fas-положительных клеток резко уменьшается и коррелирует как со снижением активности трансаминаз, так и с уменьшением выраженности портальной и лобулярной лимфоидной инфильтрации ткани печени [9]. Наконец, продуцируемый преимущественно макрофагами в избыточных количествах TNF-α также ведет к апоптозу клеток путем взаимодействия с соответствующим рецептором.
Рис. 1. Элиминация инфицированного гепатоцита цитотоксическим Т-лимфоцитом.
(ЦТЛ- цитотоксический Т-лимфоцит)
Понимание механизмов, ведущих к апоптозу клеток печени, в том числе при хроническом вирусном гепатите, позволит разработать новые методы терапии, в частности, уменьшающие избыточную гибель гепатоцитов. Одним из направлений в этой области может служить разработка ингибиторов т.н. проксимальных каспаз (2, 8, 10).
В заключение хотелось бы отметить, что, являясь универсальным биологическим механизмом, апоптоз при вирусных гепатитах может приводить к избыточной гибели не только гепатоцитов, но и других клеточных популяций, отражая либо системный иммуновоспалительный ответ на инфекцию, либо внепеченочную персистенцию вируса. В связи с этим определенный интерес представляют наши исследования апоптоза периферических лейкоцитов при хроническом гепатите. У больных хроническими гепатитами В и С и у здоровых доноров определялось количество лимфоцитов и гранулоцитов периферической крови в состоянии апоптоза непосредственно после выделения и после 24-часовой инкубации в культуральной среде методом проточной цитофлюориметрии, а также повреждение ДНК по скорости щелочной денатурации. Даннные исследования показали, что апоптоз как лимфоцитов, так и гранулоцитов был достоверно выше у больных ХГВ по сравнению с контролем как непосредственно после выделения, так и после суточной инкубации. Более интенсивный переход клеток в апоптоз наблюдался в подгруппе больных гепатитом С, хотя здесь следует учитывать роль разного количества пациентов в подгруппах (12с гепатитом В, 22 с гепатитом С). Выявлена также корреляция между степенью повреждения ДНК лимфоцитов и гранулоцитов и накоплением клеток в состоянии апоптоза после инкубации [3]. Помимо этого, после суточной инкубации количество лейкоцитов обеих популяций в апоптозе достоверно коррелировало с сывдроточной концентрацией TNF-α [4].
Полученные результаты свидетельствуют об усилении программированной гибели клеток периферической крови у больных хроническим вирусным гепатитом, что может отражать как повреждающее действие гепатотропных вирусов на лейкоциты (или их костномозговые предшественники), так и влияние системного действия провоспалительных цитокинов. Косвенным подтверждением последнего механизма служит корреляция интенсивности апоптоза с сывороточным уровнем TNF-α - наиболее мощного цитокина с проапоптогенным эффектом.
Литература
1. Аббасова С.Г., Липкин В.М., Трапезников Н.Н., Кушлинский Н.Е. Система FAS - FASL в норме и патологии // Вопр. биол., мед., фарм. химии. - 1999. - №3.-С.3-17.
2. Аруин Л.И. Апоптоз и патология печени // Росс. ж. гастроэнтерол., гепатол., колопроктол. - 1998. - №2. - С. 6-10.
3. Буеверов А.О., Тихонина Е.В., Москалева Е.Ю. и др. Апоптоз периферических лейкоцитов при хронических вирусных гепатитах // Росс. ж. гастроэнтерол., гепатол., колопроктол. - 2000. - №6. - С. 30-33.
4. Boueverov A.O., Mammaev S.N., Tikhonina E.V. et al. Enchanced apoptosis of peripheral blood leucocytes in chronic viral hepatitis // Gut. - 2000. - Vol.47 (suppl. 3). - A 181.
5. Faubion W.A., Gores G.J. Death receptors in liver biology and pathobiology // Hepatology. - 1999. - Vol.29. - P. 1-4.
6. Hayashi N., Mita E. Fas system and apoptosis in viral hepatitis//J. Gastroenterol. Hepatol. - 1997.-Vol.l2. - S.223-226.
7. Jonsson J.R., Edwards-Smith C.J., Catania S.C. et al. Expression of cytokines and factors modulating apoptosis by human sinusoidal leucocytes // J. Hepatol. - 2000. - Vol.32. -P.392-398.
8. Krams S.M., Fox S.K., Beatty P.R. et al. Human hepatocytes produce an isoform of FAS that inhibits apoptosis // Transplantation. - 1998. - Vol.65. - P.713-721.
9. Okazaki M., Hino K., Fujii К et al. Hepatic Fas-antigen expression before and after interferon therapy in patients with chronic hepatitis С // Dig. Dis. Sci. - 1996. - Vol.41. - P.2453-2458.
10. Su F, Schneider R.J. Hepatitis В virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha // Proc. Nat. Acad. Sci. USA. - 1997. - Vol.94. - P.8744-9.
Иммунный ответ эпителия кишечника на бактерии и пути активации апоптоза
В начале процесса распознавания бактерий эпителиальными клетками в течение нескольких минут активируются NFkB, МАРК и IRF3. Появление в ядре ДНК-связывающих факторов транскрипции приводит к новой транкрипции ряда эффекторных молекул. Исследования, проводимые с помощью технологии крупномасштабного профилирования экспрессии генов, позволяют выявить транскрипцию антибактериальных пептидов, цитокинов, молекул адгезии, хемотаксических мессенджеров, антиапоптозных протеинов и метаболических энзимов, которые участвуют в уничтожении бактерий и заживлении ран.
Почти все промоторы воспалительных эффекторных генов имеют необходимые для активации NFKB-связывающие локусы. Кроме того, эффекторы, задействованные на ранних фазах воспаления, часто имеют промоторсвязывающие локусы для МАРК-активированных факторов транскрипции (cJun, CREB/ATF). Сначала эти факторы транскрипции повышают экспрессию нейтрофил-специфических хемокинов и молекул адгезии лейкоцитов. Нейтрофильная инфильтрация приводит к классическим гистологическим изменениям, характерным для острого воспаления.
Эффекторы вторичной фазы часто обнаруживают IRF-связывающие локусы на своих промоторах. Эти эффекторы индуцируют появление хемокинов, факторов роста и молекул адгезии (молекула межклеточной адгезии 1, белок хемоаттрактанта моноцитов 1), необходимых для вовлечения в процесс моноцитов.
Ученые только начинают понимать механизмы специфичности иммунного ответа. Методики транскрипционного профилирования позволяют предположить, что бактериальные лиганды (ЛПС и ПГН) играют роль посредников в ответе, оптимальном для антибактериальной активности.

Пути взаимодействующих протеинов обозначены стрелками. Слева указаны функциональные классы ферментов.
ATF — трансмембранный фактортранскрипции;
IкВ — ингибитор каппа В;
IкК — IкВ-киназа;
IRAK — киназа, ассоциированная с рецептором IL-1;
IRF — фактор транскрипции, индуцирующий интерфероновый ответ;
МАМР — ассоциированные с микроорганизмами молекулярные паттерны;
МАРК — митоген-активируемая протеинкиназа;
МАРКК — киназа МАРК;
МАРККК — киназа киназы МАРК;
NFkB — нуклеарный фактор каппа В;
TLR — Toll-подобные рецепторы;
TRAF6 — ассоциированный с TNF фактор 6.
Эти компоненты, как предполагают, высвобождаются при гибели организма. Более того, в процессе воспалительной реакции происходит повышение экспрессии эндогенных провоспалительных медиаторов, таких как IL-1 или TNF, что активирует сложные вторичные паттерны генной экспрессии.
Поскольку патогены и другие стрессорные факторы активируют врожденные провоспалительные пути (например, NFkB), задействованные в процессе клеточного воспаления, они одновременно стимулируют проапоптозные пути (опосредованные каспазами), предположительно приводящие к элиминации инфицированной или необратимо поврежденной клетки. Апоптоз имеет характерные морфологические признаки и генетически контролируемый врожденный механизм, посредством которого конкретные клетки могут самоуничтожаться, максимально сохраняя при этом окружающие их клетки.
Присутствие цитохрома-с в цитоплазме определяет образование сложной структуры, называемой апоптосомой, которая необходима для активации инициирующей каспазы-9 и последующих эффекторных каспаз. В случае внешнего пути внеклеточные события, связывающие лиганды, приводят к активации комплексов рецепторов гибели клеток. Как правило, это члены семейства TNF-рецепторов (TNF-R, FAS, TRAIL и т.д.), опосредующих сборку протеинов, несущих мотив домена смерти.
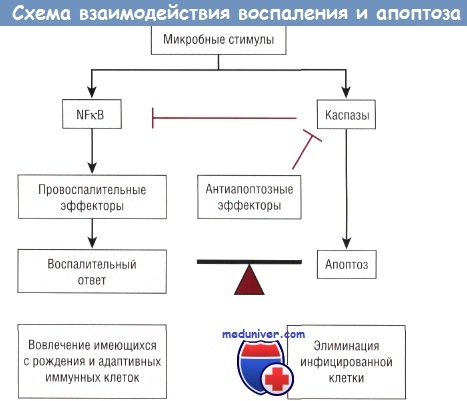
Активирующие взаимодействия обозначены стрелками, ингибирующие — линиями с ограничением.
NFkB — нуклеарный фактор каппа В.
Благодаря недавно проведенным исследованиям стало известно, что TLR и NOD-протеины могут активировать внешний путь апоптоза и использовать большую часть той же сигнальной схемы. Эти открытия положили начало возникновению парадигмы, суть которой заключается в том, что, когда клетка сталкивается с каким-либо потенциально разрушительным для нее воздействием (микробным или физического характера, как, например, гипоксия), одновременно активируются провоспалительные и проапоптозные пути. Важно установление к настоящему времени того факта, что активация NFkB (и МАРКК) представляет собой путь выживаемости клетки. Исследования, проведенные на генетических моделях у мышей, подтвердили ключевую роль NFkB в процессе антиапоптозного контроля.
Мутантные мыши, утратившие субъединицу р65 NFkB, погибают внутриутробно из-за массивного апоптоза в тканях печени. У породы мышей, имеющих только нулевые аллели IkKb в энте-роцитах кишечника, в норме не выявляются какие-либо аномалии, но в условиях системного стресса (транзиторной ишемии кишечника) отмечается массивный апоптоз энтероцитов. Фармакологическое ингибирование NFkB, потенциального проапоптозного стимулирующего фактора, используют в качестве химиотерапевтического воздействия. Почему же активация NFkB необходима для выживаемости клетки?
Помимо индуцирования NFkB провоспалительных медиаторов его активация одновременно вызывает индукцию анти-апоптозных эффекторных протеинов (например, членов семейства Al, cFLIP, Bcl, а также ингибиторов белков апоптоза), которые нужны для блокирования апоптозного сигнала на многих контрольных этапах процесса и прерывания активации каспазы. В таких условиях апоптоз останавливается и идет процесс клеточного воспаления.
С другой стороны, в случае ослабления провоспалительного сигнала под воздействием определенных стрессорных факторов активация каспазы может иметь преимущество: активированные каспазы могут действовать по принципу отрицательной обратной связи на пути выживания, разрушая сигнальных посредников воспаления. В этих условиях происходит апоптоз подверженных стрессу клеток, которые саморазрушаются (без лизиса), уничтожая чужеродные бактерии и не вызывая вторичной воспалительной реакции.
Многочисленные микробные и вирусные патогены выделяют белки-эффекторы, подавляющие активацию путей NFkB и МАРК. Именно так действует механизм иммуносупрессии и индукции апоптоза в регуляторных клетках. В контексте клеточного стресса указанные данные заставляют быть осторожными при назначении противовоспалительных препаратов.
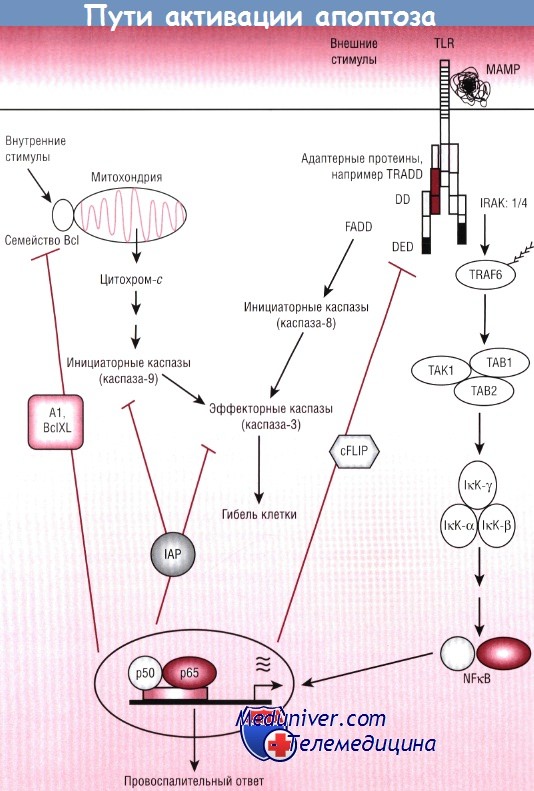
Активирующие взаимодействия обозначены стрелками, ингибирующие — линиями с ограничением.
DD — домен смерти;
DED — эффекторный домен смерти;
FADD — связанный с Fas протеин домена смерти;
IкК—IкВ-киназа;
IAP — ингибиторы белков апоптоза;
IRAK — киназа, ассоциированная с рецептором IL-1;
МАМР — ассоциированные с микроорганизмами молекулярные паттерны;
NFkB — нуклеарный фактор каппа В;
TLR — Toll-подобные рецепторы;
TRADD — связанный с TNF-рецептором протеин домена смерти;
TRAF6 — ассоциированный с TNF фактор 6.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Значение апоптоза в развитии вирусных инфекций и рака
• Вирусные инфекции и рак представляют собой такие патологические состояния, при которых может происходить блокирование апоптоза
Вирусы являются облигатными внутриклеточными паразитами, и поэтому гибель клетки-хозяина является одной из форм борьбы с вирусными инфекциями. Поэтому поддержание жизнеспособности клеток находится в интересах вируса. Многие вирусы обеспечивают это, блокируя клеточную гибель, и механизмы этой блокировки играют важную роль в понимании этапов апоптоза.
Как отмечалось, у насекомых апоптоз регулируется на уровне активации каспазы IAP. Бакуловирусы насекомых блокируют апоптоз точно таким же образом, экспрессируя IAP, который ингибирует DRONC и эффекторные каспазы. Вместе с тем, эти вирусы также экспрессируют другой белок, р35, который связывает и ингибирует активные каспазы. Блокируя активацию каспаз, эти вирусы поддерживают жизнеспособность клетки до тех пор, пока не образуется достаточное количество вирусных частиц и не произойдет лизис клетки.
Для вирусов позвоночных характерна другая стратегия, вероятно, поскольку ингибирование каспазы не блокирует повышение МОМР или независимую от каспаз гибель (и может способствовать развитию иммунного ответа на присутствие вируса). Поэтому некоторые вирусы позвоночных продуцируют антиапоптотические белки, относящиеся к семейству Вс1-2, например Е1В19К и белок BHRF вируса Эпштейна-Барра. Эти и другие вирусные Bcl-2 белки функционируют подобно Вс1-2, предотвращая повышение MOMP и апоптоз.
Вирусы позвоночных часто функционируют, предотвращая апоптоз, индуцируемый эффекторными клетками иммунной системы. Например, вирус ветряной оспы продуцирует ингибиторы протеаз, называемые серпины, которые способны блокировать гранзим В и каспазу-8 (но не каспазу-9 или эффекторные каспазы). Путем блокирования гранзима В вирус избегает эффекта цитотоксических лимфоцитов, которые ведут в организме поиск инфицированных клеток.
Аналогичным образом, ингибирование каспазы-8 приводит к блокированию апоптоза, индуцированному связыванием лигандов с рецепторами клеточной гибели; эти лиганды часто продуцируются цитотоксическими лимфоцитами и другими клетками в ответ на заражение вирусом. Еще один путь, посредством которого вирусы блокируют апоптоз с участием рецепторов клеточной гибели, связан с экспрессией молекул, близких к c-FLIP, например v-FLIP белка, продуцируемого вирусом герпеса.
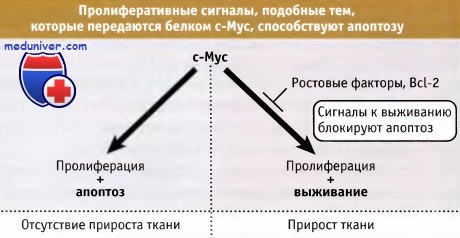
c-Myc и родственные белки не только направляют клетку в цикл, но также сенсибилизируют ее к гибели по пути апоптоза.
В результате, в отсутствие дополнительных сигналов, прироста ткани не наблюдается.
Когда апоптоз блокируется, например, ростовыми факторами, общее количество клеток начинает увеличиваться.
Способность c-Myc сенсибилизировать клетки к апоптозу является фундаментальным свойством этого белка,
а не побочным эффектом его влияния на клеточный цикл.
Подобные механизмы, вероятно, являются центральными в поддержании нормального тканевого гомеостаза и в предотвращении их злокачественного роста.
Другие патологические состояния, связанные с нарушениями механизмов апоптоза, представляют собой опухолевые заболевания. Как отмечалось в начале настоящего раздела, мощный супрессор опухолей, белок р53, который примерно в 30% случаев рака у человека представлен мутантной формой, частично оказывает свое супрессорное действие за счет индукции апоптоза в трансформированных клетках.
Аналогичным образом, антиапоптотический белок Bcl-2 впервые был обнаружен при исследовании хромосомной транслокации, происходящей при фолликулярной В-клеточной лимфоме. При раке происходят изменения в механизмах контроля апоптоза.
Однако взаимосвязь между апоптозом и раком имеет более фундаментальный характер, чем иллюстрируют эти простые примеры. Клетки позвоночных становятся чувствительными к запуску апоптоза за счет сигналов, которые обусловливают их вхождение в цикл, и принятие решения о жизни или смерти основывается на сигналах, переданных клетке от окружающих тканей (например, таких как ростовые факторы). При недостатке этих факторов, у клеток, находящихся в цикле, наступает апоптоз, и это ограничивает рост ткани. На рисунке ниже, например, показано, что белок c-Myc способствует не только пролиферации клеток, но также их апоптозу.
Поэтому экспрессия этого белка в ткани, в отсутствие других сигналов, необязательно способствует клеточной экспансии (или росту опухоли). Основные взаимоотношения между клеточным циклом и апоптозом, вероятно, имеют фундаментальное значение для выяснения вопроса о том, способны ли мы, являясь долгоживущим многоклеточным организмом, существовать без развития рака. Клеточный цикл не является причиной апоптоза; те же молекулы, которые обеспечивают вхождение клетки в цикл, также запускают и апоптоз, который подавляется антиапоптотическими сигналами.
Хотя c-Myc способствует пролиферации и апоптозу, Bcl-2 или Bcl-xL могут функционировать совместно с с-Мус и запускать процесс онкогенеза за счет блокирования апоптоза. Действительно, часто у человека при раке наблюдается активация антиапоптотических белков семейства Bcl-2. Такие кооперативные взаимоотношения между двумя типами онкогенов при раке (проапоптотические и пролиферативные, плюс антиапоптотические), вероятно, представляют собой общий принцип. В общем, можно сказать, что онкогены могут быть аналогичными c-Myc (способствующие как пролиферации, так и апоптозу), Bcl-2 (блокирующие апоптоз, не влияя на пролиферацию), или напоминать те и другие (способствующие пролиферации и блокирующие апоптоз вследствие множественности сигналов).
Белок Ras в активной форме может принимать участие в разных сигнальных путях, которые приводит к этим двум результатам. Так, в зависимости от условий, Ras может как промотировать, так и ингибировать апоптоз.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Апоптоз - функции, механизмы
Апоптозом называется запрограммированная клеточная гибель. Этот процесс играет важную роль в росте и развитии организма, т. к. по мере созревания тканей некоторые клетки должны погибнуть, чтобы уступить место более дифференцированным и специализированным.
Если клетка не умирает и становится бессмертной, может развиться злокачественная опухоль. Впервые апоптоз был описан в 1970-е годы, но только в последнее время исследователи осознали его важную роль для развития организма, дифференцировки тканей и малигнизации.
Интерес к апоптозу возрос, когда выяснилось, что этот процесс находится под контролем генов, вовлеченных в злокачественную трансформацию, т. е. онкогенов, протоонкогенов и генов-супрессоров. Очевидно, что многие из этих генов активны во время развития организма.
Полагают, что изучение апоптоза и путей его регуляции позволит понять механизмы развития организма и старения. Утрата клеточного контроля над программированной клеточной гибелью ведет к развитию опухолей.
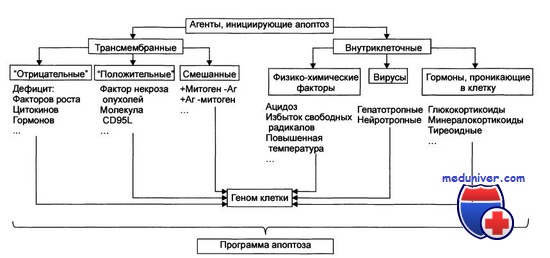
Стадия инициации апоптоза
Апоптоз — особый вид клеточной гибели, ответственный за устранение клеток в нормальных тканях. Тем не менее этот процесс наблюдается и при патологических процессах. Гистологически проявляется уменьшением клетки, буллезным разрушением клеточной мембраны и конденсацией клеточного ядра.
В итоге образуются апоптотические тельца, содержащие неповрежденные органеллы; окружающие клетки фагоцитируют эти тельца. Апоптоз не сопровождается воспалением, что отличает его от некроза. Последний сопровождается набуханием клетки, разрушением всех ее структур и развитием воспалительного ответа.
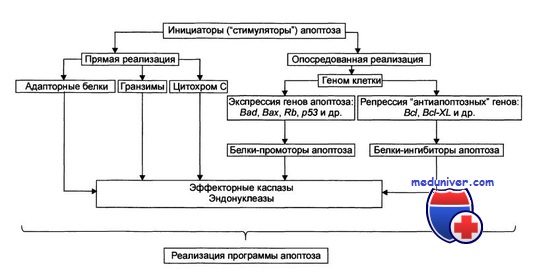
Стадия программирования апоптоза
Апоптоз играет важную роль в нормальном росте организма, а также в развитии и прогрессировании злокачественных опухолей. Спонтанный апоптоз встречается в злокачественных клетках и даже замедляет их рост.
Интенсивность этого процесса возрастает при облучении опухоли, проведении гормоно- и химиотерапии, при нагревании опухоли. В злокачественных опухолях апоптоз представляет механизм уничтожения клеток, в которых произошли канцерогенные изменения ДНК.
Однако если он заблокирован или подавлен мутациями контролирующих его генов, например BCL2 или ТР53, то эти клетки получают возможность свободно делиться и неограниченно накапливать мутации. Такая генетическая нестабильность — ранний этап развития злокачественных опухолей.
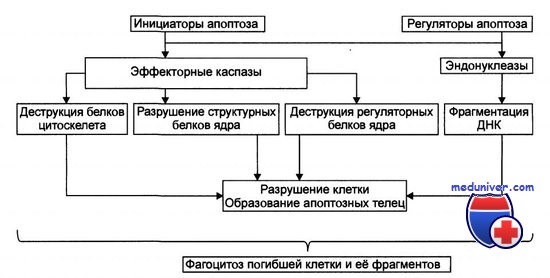
Стадия реализации апоптоза
Многие из современных методов лечения, например лучевая и химиотерапия, направлены на уничтожение клеток за счет повреждения их ДНК. Мутации гена BCL2 или ТР53 ухудшают эффективность лечения, т. к. подавляют клеточную гибель.
Более глубокое понимание процессов запрограммированной клеточной гибели может привести к разработке новых, более эффективных методов лечения. Ингибиторы апоптоза (например, протоонкоген BCL2) могут быть ответственны за развитие резистентности к противоопухолевым препаратам, позволяя выживать клеткам с патологической ДНК.
Вероятно, в дальнейшем будут выявлены и другие механизмы подавления апоптоза. Не следует думать, что этот процесс отражает нечто иллюзорное в биологической литературе, а термин принят только для описания отличной от некроза клеточной гибели. Апоптоз — фундаментальный процесс, контролируемый на молекулярном уровне, и можно надеяться, что его удастся расшифровать и использовать для медицинских нужд. Возможные механизмы апоптоза представлены на рисунке.
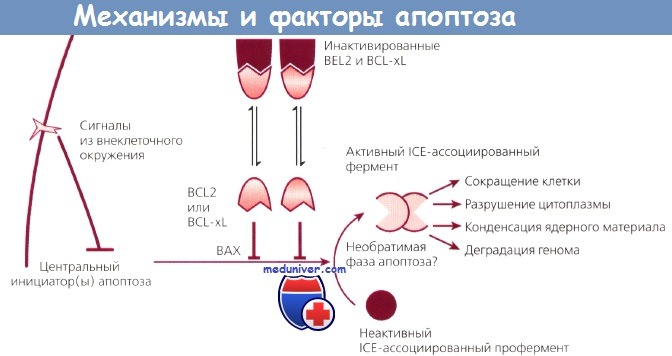
Возможные механизмы апоптоза и факторы, его контролирующие.
Внеклеточный сигнал запускает каскад событий, вовлекающий молекулы BCL2, BCL-xL и ВАХ.
Это ведет к наступлению программированной гибели клетки.
Этот механизм может быть заблокирован на любом из множества этапов, в результате чего клетка становится бессмертной.
ICE - интерлейкин-1b-превращающий фермент.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Обследованы 78 часто болеющих детей в возрасте от 3 до 6 лет с хронической Эпштейна-Барр вирусной инфекцией. Микст-инфекция вируса Эпштейна-Барр с цитомегаловирусом и/или вирусом простого герпеса характеризовалась высокой частотой факторов риска в антенатальном и постнатальном периодах, бактериально-грибковых микст-инфекций, поражения нижних дыхательных путей и легких, проявлений лимфопролиферативного, церебрального и гастроинтестинального синдромов, глубокими нарушениями Т-клеточного, В-клеточного звеньев иммунитета, метаболизма нейтрофилов. У пациентов с хронической Эпштейна-Барр моноинфекцией обнаружены поражение верхних дыхательных путей, выраженные кардиальный и артралгический синдромы, активация В-клеточного звена иммунитета. Лечение с использованием инозина пранобекс (изопринозина) и рекомбинантного интерферона α2b (виферона) приводило к торможению репликации герпесвирусов, модуляции показателей клеточного и гуморального иммунного ответа, нормализации метаболической активности нейтрофилов, уменьшению частоты респираторных инфекций и симптомов хронической Эпштейна-Барр вирусной инфекции.
Проблема часто болеющих детей (ЧБД) по-прежнему сохраняет свою высокую актуальность. Группа ЧБД - это группа диспансерного наблюдения, которая включает пациентов с частыми респираторными инфекциями, возникающими из-за транзиторных, корригируемых отклонений в защитных системах организма, и не имеющих стойких органических нарушений в них [1, 2]. К причинам формирования группы ЧБД относят анатомо-физиологические особенности детского организма, в том числе иммунной системы, развитие вторичного иммунодефицитного состояния (ИДС), увеличение частоты контактов с больными при посещении организованных коллективов и др. 3. К группе часто болеющих относятся 40% детей дошкольного возраста и 15% учащихся младших классов [1-3, 5]. Высокая частота инфекционных заболеваний респираторного тракта приводит к ряду неблагоприятных последствий -нарушению физического и нервно-психического развития, задержке созревания иммунной системы, формированию хронической патологии ЛОР-органов, желудочно-кишечного тракта, мочевыделительной и других систем, социальной дезадаптации ребенка [1-3, 5, 6]. В последнее время установлена значительная распространенность оппортунистических инфекций (герпесвирусных инфекций, хламидиоза, микоплазмоза) у данной категории пациентов, что диктует необходимость разработки новых подходов к их лечению 7. Перспективным направлением совершенствования комплексной терапии ЧБД представляется включение в программу лечения инозина пранобекс (изопринозина), который обладают противовирусной, иммунокорригирующей и цитопротективной активностью [9, 10].
Цель работы - оптимизация программы лечения ЧБД с использованием инозина пранобекс (изопринозина).
Материалы и методы исследования
Под нашим наблюдением находились 78 пациентов в возрасте от 3 до 6 лет, относящихся к группе ЧБД. Частота эпизодов острых респираторных инфекций (ОРИ) составляла 6-12 раз в год. У всех больных на основании клинико-лабораторного обследования диагностирована хроническая Эпштейна-Барр вирусная инфекция (ХЭБВИ) в стадии реактивации, которая у 28 детей (1-я группа; 35,9%) протекала в форме моно-инфекции, у 50 больных (2-я группа, 64,1%) - в виде микст-инфекции в сочетании с другими герпесвирусными инфекциями, в том числе с цитомегаловирусной инфекцией (ЦМВИ) и инфекцией простого герпеса (ИПГ). Методом случайной выборки больные были распределены на 2 группы, оказавшиеся сопоставимыми по клинико-лабораторным показателям на момент начала лечения. Всем пациентам назначали рекомбинантный интерферон α2b (ИФНα2b, виферон) по пролонгированной схеме [11], пробиотики, препараты метаболической реабилитации и поливитамины. В комплексную терапию 40 пациентов (15 детей 1-й группы и 25 больных 2-й группы) был включен инозин пранобекс (изопринозин) в дозе 50-100 мг/кг/сут в 3-4 приема внутрь. Проводили 3 курса лечения по 10 дней с интервалом 10 дней. Остальные дети (13 пациентов 1-й группы и 25 больных 2-й группы) получали только стандартную терапию.
Результаты и их обсуждение
Изучение преморбидного фона выявило его отягощенность у всех обследованных детей. Причем, у пациентов с герпесвирусной микст-инфекцией достоверно чаще, чем у больных 1-й группы, регистрировались факторы риска у матерей (соматические заболевания - 44% и 21,4%, отягощенный акушерский анамнез, в том числе гестоз -58% и 35,7%, угроза прерывания беременности -64% и 39,3%, гипоксия плода - 72% и 50%, инфекционные заболевания во время беременности -66% и 32,1%, преждевременные роды - 20% и 3,6%) и у детей (перинатальное поражение ЦНС -82% и 60,7%, врожденные пороки развития -32% и 10,7%, асфиксия - 52% и 28,6%, задержка внутриутробного развития - 20% и 3,6%, лимфатико-гипопластический диатез - 42% и 17,8%, пневмонии - 36% и 14,3%, атопический дерматит - 30% и 10,7%) (р<0,05).
Синдромальная модель ХЭБВИ у детей обеих групп включала признаки хронической интоксикации (длительный субфебрилитет, слабость, снижение аппетита, раздражительность и др.) у 78,6% детей 1-й группы и 100% детей 2-й группы и лимфопролиферативный синдром - генерализованная лимфаденопатия (ГЛАП) у всех детей обеих групп, гипертрофия небных (92,9% и 92%) и глоточной 957,1% и 60%) миндалин, увеличение печени (25% и 52%) и селезенки (7,1% и 26%). Проявлениями инфекционного синдрома являлись повторные острые инфекции респираторного тракта и ЛОР-органов у всех детей. Достаточно часто имели место изменения со стороны ЦНС в виде гипертензионно-гидроцефального (21,4% и 54%), вегето-висцерального (57,1% и 34%) синдромов и синдрома гиперактивности (53,6% и 54%), патология органов пищеварения (аномалии развития - 17,9% и 40%, хронический гастрит -39,3% и 44%, дискинезия желчевыводящих путей - 50% и 28%), сердечно-сосудистой системы (боли в сердце, сердцебиение - у 42,9% и 22%) и артралгии (28,6% и 10% соответственно).
При герпесвирусной микст-инфекции чаще, чем у пациентов 1-й группы, регистрировались симптомы интоксикации, увеличение печени и селезенки, гипертензионно-гидроцефальный синдром, аномалии развития желудочно-кишечного тракта. У этих пациентов отмечалась более высокая, чем при моно-инфекции ВЭБ, частота эпизодов ОРИ (9,5±1,1 раз и 6,8±1,2 раз в год соответственно; р<0,05), которые часто осложнялись развитием бронхита (60% и 25% соответственно; р<0,05) и пневмонии (30% и 10,7% соответственно; р<0,05).
Особенностью моно-инфекции ВЭБ являлись более частое развитие вегето-висцерального синдрома, дискинезии желчевыводящих путей, кардиального и артралгического синдромов. ОРИ у этих больных протекали преимущественно в форме фаринготонзиллита, аденоидита или ринофарингита.
Таблица 1
Показатели иммунного статуса ЧБД с хронической Эпштейна-Барр вирусной инфекцией
| Показатели | 1-я группа | 2-я группа | Здоровые дети |
| CD3, % | 67,2±1,2 1 | 67,6±1,3 1 | 74,5±1,9 |
| CD4, % | 43,1±1,1 1,2 | 39,1±1,1 1 | 49,5±1,9 |
| CD8, % | 26,8±1,2 1,2 | 24,1±1,1 1 | 21,5±1,1 |
| CD4/CD8 | 1,6±0,1 1 | 1,6±0,1 1 | 2,3±0,1 |
| CD25, % | 5,9±0,5 1,2 | 3,5±0,6 1 | 4,8±0,4 |
| HLA DR, % | 15,5±1,1 1,2 | 13,1±1,2 1 | 4,5±0,2 |
| CD95, % | 6,1±0,5 1,2 | 7,2±0,4 1 | 3,5±0,4 |
| CD20, % | 11,2±1,3 1,2 | 14,9±1,2 1 | 20,4±0,4 |
| IgA, г/л | 1,31±0,1 1,2 | 1,1±0,05 1 | 0,8±0,03 |
| IgM, г/л | 1,65±0,1 1,2 | 1,36±0,1 1 | 0,86±0,1 |
| IgG, г/л | 11,5±0,2 1,2 | 10,1±0,2 1 | 8,7±0,4 |
| ЦИК, усл. ед. | 143,4±6,7 1,2 | 73,7±6,8 1 | 43,2±4,4 |
| CD16, % | 12,8±1,1 2 | 11,1±0,8 1 | 12,7±0,6 |
| НСТ сп., усл. ед. | 123,4±5,2 1,2 | 112,1±5,1 1 | 100,1±5,1 |
| К ст. НСТ | 1,7±0,1 1,2 | 1,5±0,05 1 | 1,9±0,05 |
р
Сочетанное использование инозина пранобекс (изопринозина) и рекомбинантного ИФНα2b (виферона) у больных моноинфекцией ВЭБ способствовало отчетливой положительной динамике клинических показателей, в том числе частоты ГЛАП (до лечения 100%, после лечения 60%), гипертрофии небных и глоточной миндалин (53,3% и 13,2%), гепатомегалии (26,7% и 0%), интоксикационного (73,3% и 33,3%), инфекционного (100% и 60%), вегето-висцерального (53,3% и 13,3%) синдромов, синдрома гиперактивности (60% и 20%), дискинезии желчевыводящих путей (46,6% и 13,3%), кардиального (46,6% и 13,3%) и артралгического (33,3% и 0%) синдромов. У большинства пациентов исчезали серологические показатели репликации ВЭБ. При назначении монотерапии рекомбинантным ИФНα2b (вифероном) отмечалась лишь тенденция к уменьшению указанных показателей. Частота эпизодов ОРИ в течение 3 месяцев у детей, получавших рекомбинантный ИФНα2b (виферон), составила 1,9±0,3 раз, на фоне комбинированной терапии - 0,8±0,2 раз (р<0,05).
Таблица 2
Показатели иммунного статуса ЧБД с хронической Эпштейна-Барр вирусной моноинфекцией с учетом программы лечения
| Показатели | Больные, получавшие изопринозин и виферон (n=15) | Больные, получавшие виферон (n=13) | Здоровые дети | ||
| до лечения | после лечения | до лечения | после лечения | ||
| CD3, % | 63,8±1,4 1,2 | 71,3±1,4 3 | 66,7±1,3 1 | 68,2±1,3 1 | 74,5±1,9 |
| CD4, % | 44,1±1,2 1,2 | 46,9±1,2 3 | 42,1±1,2 1 | 44,2±1,3 1 | 49,5±1,9 |
| CD8, % | 25,7±1,3 1,2 | 28,4±1,2 1,3 | 27,3±1,3 1 | 25,7±1,2 1 | 21,5±1,1 |
| CD4/CD8 | 1,7±0,1 1 | 1,6±0,1 1 | 1,5±0,1 1 | 1,7±0,1 1 | 2,3±0,1 |
| CD25, % | 5,8±0,5 1,2 | 6,9±0,4 1,3 | 6,3±0,6 1 | 5,9±0,4 1 | 4,8±0,4 |
| HLA DR, % | 15,1±1,1 1,2 | 17,5±1,1 1,3 | 15,9±1,2 1 | 16,1±1,1 1 | 4,5±0,2 |
| CD95, % | 5,7±0,6 1,2 | 4,4±0,5 3 | 6,3±0,6 1 | 5,3±0,3 1 | 3,5±0,4 |
| CD20, % | 12,2±1,3 1,2 | 15,8±1,2 1,3 | 10,8±1,3 1,2 | 13,4±1,2 1 | 20,4±0,4 |
| IgA, г/л | 1,25±0,1 1,2 | 1,5±0,05 1,3 | 1,36±0,1 1 | 1,38±0,06 1 | 0,8±0,03 |
| IgM, г/л | 1,6±0,1 1,2 | 1,1±0,2 3 | 1,7±0,1 1,2 | 1,5±0,08 1 | 0,86±0,1 |
| IgG, г/л | 11,4±0,3 1,2 | 12,3±0,3 1,3 | 11,7±0,3 1 | 11,3±0,2 1 | 8,7±0,4 |
| ЦИК, усл. ед. | 148,7±7,8 1,2 | 84,7±6,5 1,3 | 141,4±7,1 1,2 | 126,3±8,4 1 | 43,2±4,4 |
| CD16, % | 11,8±1,2 2 | 14,4±1,2 1,3 | 13,6±1,3 | 11,8±1,2 | 12,7±0,6 |
| НСТ сп., усл. ед. | 121,6±5,6 1,2 | 137,4±6,1 1,3 | 125,1±5,8 1 | 124,8±5,9 1 | 100,1±5,1 |
| К ст. НСТ | 1,7±0,1 1,2 | 1,9±0,06 3 | 1,7±0,1 1 | 1,7±0,1 1 | 1,9±0,05 |
Здесь и в табл. 3: р
У пациентов 2-й группы на фоне комбинированного лечения, наблюдалась более существенная, по сравнению с монотерапией, динамика клинических показателей - снижение частоты лимфопролиферативного (100% и 68%), интоксикационного 9100% и 44%), инфекционного (100% и 68%), церебрального, гастроинтестинального, кардиального (24% и 4%) и артралгического (12% и 4%) синдромов. При использовании монотерапии рекомбинантным ИФНα2b (вифероном) частота эпизодов ОРИ в течение 3 месяцев составила 2,1±0,4 раз, при назначении комбинированного лечения - 1,1±0,2 раз (р<0,05). Кроме того, у детей, получавших сочетание инозина пранобекс (изопринозина) и рекомбинантного ИФНα2b (виферона), в отличие от монотерапии, отмечалось существенное уменьшение частоты серологических маркеров репликации ВЭБ, ЦМВ и ВПГ.
Таблица 3
Показатели иммунного статуса ЧБД с хронической Эпштейна-Барр микствирусной инфекцией с учетом программы лечения
| Показатели | Больные, получавшие изопринозин и виферон (n=25) | Больные, получавшие виферон (n=25) | Здоровые дети | ||
| до лечения | после лечения | до лечения | после лечения | ||
| CD3, % | 66,7±1,4 1,2 | 70,1±1,3 1,3 | 67,9±1,4 1 | 67,4±1,2 1 | 74,5±1,9 |
| CD4, % | 38,7±1,2 1,2 | 44,8±1,4 1,3 | 40,3±1,2 1 | 41,4±1,3 1 | 49,5±1,9 |
| CD8, % | 23,6±1,2 1,2 | 28,6±1,3 1,3 | 24,8±1,3 1 | 25,6±1,3 1 | 21,5±1,1 |
| CD4/CD8 | 1,6±0,1 1 | 1,6±0,1 1 | 1,6±0,1 1 | 1,6±0,1 1 | 2,3±0,1 |
| CD25, % | 3,3±0,7 1,2 | 4,9±0,7 3 | 3,6±0,7 1 | 3,7±0,4 1 | 4,8±0,4 |
| HLA DR, % | 12,8±1,3 1,2 | 15,6±1,3 1,3 | 13,5±1,3 1 | 13±1,1 1 | 4,5±0,2 |
| CD95, % | 7,4±0,5 1,2 | 4,3±0,6 1,3 | 7,1±0,5 1,2 | 5,8±0,6 1 | 3,5±0,4 |
| CD20, % | 14,6±1,3 1,2 | 18,4±1,2 1,3 | 15,1±1,3 1 | 16,2±1,3 1 | 20,4±0,4 |
| IgA, г/л | 1±0,07 1,2 | 1,4±0,06 1,3 | 1,2±0,08 1 | 1,28±0,06 1 | 0,8±0,03 |
| IgM, г/л | 1,4±0,1 1,2 | 1,1±0,1 3 | 1,3±0,1 1 | 1,3±0,06 1 | 0,86±0,1 |
| IgG, г/л | 9,8±0,3 1,2 | 11,3±0,4 1,3 | 10,3±0,3 1 | 10,1±0,3 1 | 8,7±0,4 |
| ЦИК, усл. ед. | 71,3±7,2 1,2 | 53,7±5,1 1,3 | 79,3±7,7 1,2 | 54,1±4,2 1 | 43,2±4,4 |
| CD16, % | 10,8±1 1,2 | 14,3±0,6 1,3 | 11,8±0,9 | 12,8±0,8 | 12,7±0,6 |
| НСТ сп., усл. ед. | 111,6±5,3 1,2 | 126,4±5,2 1,3 | 114,7±5,6 1 | 115,4±4,2 1 | 100,1±5,1 |
| К ст. НСТ | 1,4±0,1 1,2 | 1,8±0,1 3 | 1,6±0,1 1 | 1,6±0,1 1 | 1,9±0,05 |
Здесь и в табл. 3: р
Побочные эффекты при использовании инозина пранобекс (изопринозина) отсутствовали.
Результаты проведенного исследования свидетельствуют о важной роли, которую играет хронические формы герпесвирусных инфекций в формировании группы ЧБД (рис. 1). Действие комплекса неблагоприятных факторов в антенатальном и постнатальном периодах, нарушение нейро-иммуноэндокринной регуляции гомеостаза, генетическая предрасположенность приводят к развитию фонового вторичного ИДС [4, 8]. С учетом характера и степени выраженности последнего при инфицировании ребенка герпесвирусами происходит формирование хронических форм герпесвирусных моно- или микст-инфекций (ВЭБ, ВЭБ + ЦМВ, ВЭБ + ВПГ, ВЭБ + ВПГ + ЦМВ). В результате мощного иммуносупрессивного действия герпесвирусов происходит прогрессирование иммунологических нарушений [9, 12-14]. При моноинфекции ВЭБ отмечается преимущественная активация В-клеточного звена иммунной системы, при герпесвирусной микст-инфекции - угнетение иммунного ответа по клеточному и гуморальному типам, нарушение функциональной активности факторов врожденной резистентности. Изменения со стороны местного и системного иммунитета приводят к возникновению бактериально-грибковых и хламидийно-микоплазменных ассоциаций, что особенно часто имеет место при сочетанной герпесвирусной инфекции. Одной из клинических манифестаций хронических герпесвирусных инфекций и индуцируемого ими ИДС являются повторные эпизоды острых инфекций органов дыхания. Причем, при герпесвирусной микст-инфекции инфекционный синдром выражен в большей степени, характеризуется поражением нижних отделов респираторного тракта и легких. Кроме того, у ЧБД с хронической герпесвирусной инфекцией имеет место полиорганная патология, связанная с основным заболеванием. Регистрируются признаки хронической интоксикации, лимфопролиферативный синдром (ГЛАП, гипертрофия небных и глоточной миндалин, увеличение печени и селезенки), патология со стороны ЦНС, желудочно-кишечного тракта, сердечно-сосудистой и костно-мышечной систем. При герпесвирусной микст-инфекции проявления лимфопролиферативного, интоксикационного, церебрального и гастроинтестинального синдромов встречаются чаще. Особенностью моно-инфекции ВЭБ является высокая частота вегето-висцерального синдрома, дискинезии желчевыводящих путей, кардиального и артралгического синдромов, что связано, по-видимому, запуском аутоиммунных реакций в условиях активации В-лимфоцитов 14.
Рис. 1. Иммунопатогенез формирования группы часто болеющих детей с хроническими герпесвирусными инфекциями.
Рис. 2. Механизмы этиопатогенетического действия комбинации инозина пранобекс (изопринозина) и рекомбинантного интерферона- α2b (виферона) у часто болеющих детей с хроническими герпесвирусными инфекциями.
Выводы
1. В формировании группы ЧБД важную роль играют хронические герпесвирусные инфекции, которые приводят к развитию вторичного ИДС с преимущественным повреждением В-клеточного звена при моноинфекции ВЭБ или с сочетанным нарушением иммунного статуса при микст-инфекции (ВЭБ + ЦМВ, ВПГ).
2. Прогрессирование иммуносупрессии ведет к возникновению полиорганной патологии, которая включает инфекционный, интоксикационный, лимфопролиферативный, церебральный, гастроинтестинальный, кардиальный, артралгический синдромы и выражена в большей степени при микст-инфекции.
3. Сочетанное пролонгированное назначение инозина пранобекс (изопринозина) и рекомбинантного ИФНα2b (виферона) приводит к торможению репликации герпесвирусов, восстановлению нарушенных показателей иммунного статуса, уменьшению частоты инфекционного синдрома и других клинических проявлений.
4. Высокая эффективность предложенной программы терапии позволяет рекомендовать ее включение в стандарт лечения и реабилитации ЧБД с хроническими герпесвирусными инфекциями.
Читайте также: