
Протоонкогены отличаются от вирусных онкогенов
Обновлено: 19.04.2024
Механизмы действия онкогенов и опухолевых супрессоров.
Обзор
Канцерогенез - многоступенчатый процесс накопления мутаций и других генетических изменений, приводящих к нарушениям ключевых клеточных функций, таких как регуляция пролиферации и дифференцировки, естественной гибели клеток (апоптоз), морфогенетических реакций клетки, а также, вероятно, к неэффективному функционированию факторов специфического и неспецифического противоопухолевого иммунитета. Только совокупность таких изменений, приобретаемая, как правило, в результате довольно длительной эволюции неопластических клонов, в ходе которой происходит отбор клеток с необходимыми признаками, может обеспечить развитие злокачественного новообразования. Вероятность возникновения в одной клетке нескольких генетических изменений резко повышается при нарушениях работы систем, контролирующих целостность генома. Поэтому мутации, ведущие к генетической нестабильности, также являются неотъемлемым этапом опухолевой прогрессии. Более того, некоторые врожденные аномалии систем генетического контроля являются фактором, предопределяющим неизбежное возникновение новообразования: они настолько увеличивают вероятность появления в каждой клетке организма различных онкогенных мутаций, что у индивидуума раньше или позже в какой-то из клеток пролиферирующего клона под давлением отбора обязательно накопится необходимая совокупность изменений и образуется опухоль.
Значительный прогресс в понимании механизмов канцерогенеза связан с открытием сначала онкогенов и протонкогенов, а затем - опухолевых супрессоров и мутаторных генов. Онкогены - это клеточные или вирусные (вносимые вирусом в клетку) гены, экспрессия которых может привести к развитию новообразования. Протоонкогены - нормальные клеточные гены, усиление или модификация функции которых превращает их в онкогены. Опухолевые супрессоры (антионкогены, рецессивные опухолевые гены) - клеточные гены, инактивация которых резко увеличивает вероятность возникновения новообразований, а восстановление функции, наоборот, может подавить рост опухолевых клеток. Следует заметить, что причисляемые к опухолевым супрессорам так называемые "мутаторные" гены, т.е. гены, нарушения функции которых тем или иным способом увеличивает темп возникновения мутаций и/или других генетических изменений, могут и не влиять на рост неопластических клеток. Однако их инактивация столь сильно увеличивает вероятность появления различных онкогенных мутаций, что образование опухоли становится лишь делом времени.
Принадлежность к онкогенам или опухолевым супрессорам определяется несколькими критериями: а) закономерным характером изменений структуры и/или экспрессии данного гена в клетках определенных или различных новообразований; б) возникновением в юном или молодом возрасте определенных форм опухолей у индивидов с передающимися по наследству герминальными (т.е. произошедшими в половой клетке) мутациями данного гена; в) резким повышением частоты появления опухолей у трансгенных животных, либо экспрессирующих активированную форму данного гена - в случае онкогенов, либо несущих инактивирующие мутации ("нокаут") данного гена - в случае опухолевых супрессоров; г) способностью вызывать в культивируемых in vitro клетках морфологическую трансформацию и/или неограниченный рост (онкогены), либо подавление клеточного роста и/или выраженности признаков трансформации (опухолевые супрессоры).
Два последних десятилетия характеризовались бурным открытием все новых и новых онкогенов и опухолевых супрессоров. К настоящему времени известно около сотни потенциальных онкогенов (клеточных и вирусных) и около двух десятков опухолевых супрессоров. Были описаны генетические события, приводящие к активации протоонкогенов или инактивации опухолевых супрессоров 1. Обнаружено, что механизм действия вирусных онкогенов связан с активацией клеточных протоонкогенов (ретровирусы) или инактивацией опухолевых супрессоров (ДНК-содержащие вирусы) 7. Выявлены характерные для тех или иных форм новообразований человека изменения онкогенов и опухолевых супрессоров, в том числе высокоспецифичные аномалии, используемые для постановки диагноза [3,12] (табл. 1, 2).
Таблица 1.
Некоторые изменения протоонкогенов, характерные для новообразований человека
*Подчеркнуты наследственные формы заболеваний, возникающие при мутациях в половых клетках. В остальных случаях мутации происходят в соматических клетках, которые образуют опухоли.
Таблица 2.
Формы опухолей человека, возникающие при инактивации некоторых опухолевых супрессоров и мутаторных генов
*Подчеркнуты наследственные формы заболеваний, возникающие при мутациях в половых клетках.
** Локус INK4a/ARF кодирует два белка: p16 INK4a - ингибитор циклинзависимых киназ Cdk4/6 и p19 ARF (Alternative Reading Frame) - продукт альтернативной рамки считывания, который, связывая р53 и Mdm2, блокирует их взаимодействие и препятствует деградации р53 [13, 14]. Делеции и многие точечные мутации в локусе INK4a/ARF вызывают одновременно инактивацию супрессорных активностей обоих этих белков [15].
Однако долгое время знания о каждом из онкогенов или опухолевых супрессоров представлялись дискретными, в значительной мере не связанными между собой. И лишь в самые последние годы стала вырисовываться общая картина, показывающая, что подавляющее большинство известных протоонкогенов и опухолевых супрессоров являются компонентами нескольких общих сигнальных путей, контролирующих клеточный цикл, апоптоз, целостность генома, морфогенетические реакции и дифференцировку клеток. Очевидно, изменения именно в этих сигнальных путях в конце концов и приводят к развитию злокачественных новообразований. Далее приведены сведения об основных мишенях действия онкогенов и опухолевых супрессоров.
Онкогены и их функциональная классификация
За последние 25 лет произошел существенный прогресс в нашем понимании молекулярных механизмов развития злокачественных опухолей. Идентифицировано три типа генов, нарушения в которых приводят к раку: доминантные трансформирующие, или онкогены; рецессивные трансформирующие, или опухолевые супрессоры; гены, ответственные за репарацию ДНК. Многие онкогены были впервые выделены как формы протоонкогенов онкогенных РНК-содержащих вирусов.
На протяжении многих лет известно, что вирусы способны вызывать злокачественные опухоли у животных. Это наблюдение послужило стимулом к обширным исследованиям, направленным на выявление генов, которые вызывают рак и переносятся вирусами, и генов человека, повреждающихся при развитии злокачественных опухолей. В итоге обнаружили удивительный факт, что гены, вовлеченные в канцерогенез, часто представляют собой измененные формы вирусных генов.
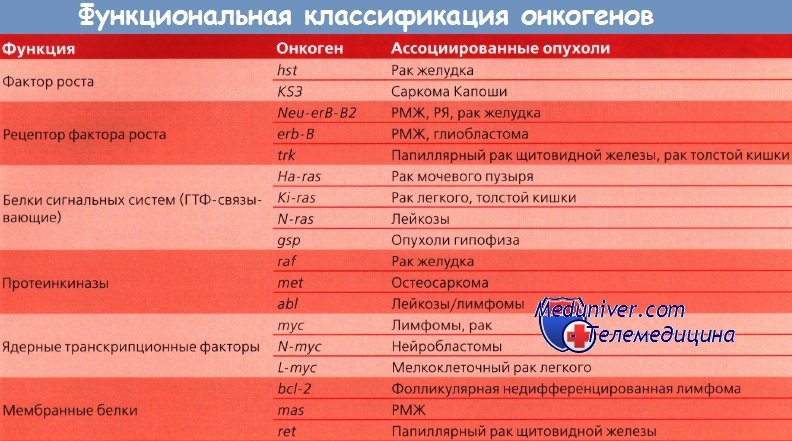
Другие протоонкогены кодируют внутриклеточные белки, ответственные за усиление митогенного сигнала; третьи — кодируют белки, вовлеченные в контроль клеточного деления и находящиеся под контролем ядра. Такие онкогены могут быть активированы посредством нескольких механизмов: возможна амплификация гена и активация его копий; в редких случаях наблюдается траислокация гена на другую хромосому, где он попадает под контроль чужого промотора и стимулирует неконтролируемый рост. Три группы онкогенов находятся под разным контролем.
Первая группа включает пептидные факторы роста и их рецепторы, как, например, эпидермальный или тромбоцитарный.
Эти пептиды служат скорее костимуляторами опухолевого роста, чем факторами, непосредственно инициирующими опухолевую трансформацию. По мере расшифровки механизмов, стимулирующих опухолевую прогрессию, все более реальной становится молекулярно-нацеленная (таргетная) терапия, направленная непосредственно против продуктов этих генов или против белков, активируемых ростовыми факторами. Так разработаны препараты:
1) трастузумаб (Герцептин) — моноклональные антитела, блокирующие Her-2/neu;
2) иматиниба мезилат (Гливек) — препарат, частично блокирующий активность c-kit, BCR-ABL и некоторых других тирозинкиназ;
3) цетуксимаб (Эрбитукс) — моноклональные антитела, связывающиеся с рецептором эпидермального фактора роста;
4) гефитиниб (Пресса) — низкомолекулярный ингибитор некоторых изоформ рецептора эпидермалыюго фактора роста.

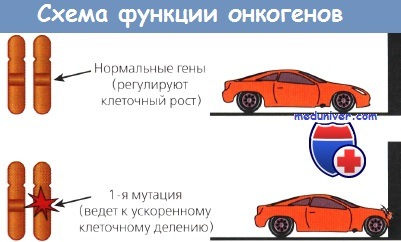
Другой класс онкогенов происходит из немембранных внеядерных факторов роста, передающих каскадные сигналы внутри клетки. К ним относятся G-белки и белки семейства ras. Наконец, некоторые онкогены кодируют ядерные регуляторные белки, например myc. Схематическое изображение функций онкогенов представлено на рисунке.
Открытие человеческих аналогов вирусных генов привело к формулировке многообещающей гипотезы, согласно которой злокачественные опухоли человека, включая большинство невирусной этиологии, могут возникать вследствие мутаций, превращающих полезные протоонкогены в опасные онкогены. В подтверждение этой гипотезы показано, что повреждение даже одного аллеля таких протоонкогенов достаточно для злокачественной трансформации некоторых клеток in vitro. Подобные доминантные мутации ведут к гиперэкспрессии нормального гена и, следовательно, гиперпродукции нормального белка или же к синтезу его аберрантной формы, обладающей повышенной активностью.
В любом случае результатом оказывается усиление стимулирующих сигналов внутри клетки, даже в отсутствие стимулирующих воздействий извне.
Парадоксально, но первым ключом к выявлению некоторых онкогенов стало исследование РНК-содержащих онкогенных вирусов животных, не вызывающих опухолей у человека. Эти ретровирусы, инфицирующие кур, грызунов, кошек и обезьян, оказались крайне высокоонкогенными: развитие опухоли часто наблюдали уже при первом контакте. У одного из вирусов этой группы, вируса саркомы кур Рауса, выявлен ген, ответственный за злокачественное перерождение инфицированных клеток. Этот тип трансформирующих онкогенов был назван вирусным онкогеном.
Единственный онкоген вируса саркомы Рауса, проникая в клетки кур, способен нарушать и перестраивать их метаболизм, направляя его по пути злокачественной трансформации.
В 1976 г. Varmus и Bishop показали, что онкоген вируса саркомы Рауса в действительности вовсе не есть вирусный ген, а происходит от предсуществовавшего клеточного гена, захваченного предком вируса саркомы Рауса. Однажды встроив его в свой геном, вирус далее использовал этот ген для трансформации клеток млекопитающих.
Более ранние предки вируса саркомы Рауса были способны реплицироваться в инфицированных клетках, но не могли трансформировать ее; туморогенный потенциал был приобретен после захвата нормального клеточного гена — протоонкогена. В связи с этим значение работы Varmus и Bishop гораздо больше, чем просто исследование вируса саркомы Рауса: показано существование гена в нормальном геноме клеток млекопитающих, обладающем трансформирующим потенциалом при соответствующей активации, в данном случае ретровирусом.
Информация, полученная при исследовании ретровирусов и онкогенов, существенно помогла в изучении причин злокачественных новообразований. Ретровирусы, так же как вирус саркомы Рауса, неинфекционны для человека и, соответственно, не могут активировать протоонкогены человека. Однако возможны альтернативные механизмы их активации. Эффекты, сходные с производимыми вирусом в последовательности ДНК, могут быть вызваны химическими или физическими воздействиями.
Это было подтверждено в начале 80-х годов прошлого столетия: в геноме опухолевых клеток были выявлены мутированные гены (протоонкогены). Во всех случаях причиной превращения протоонкогена в активный онкоген оказались изменения последовательности гена. Например, онкоген ras образуется из протоонкогена-предшественника в клетках рака мочевого пузыря человека вследствие замены одной пары нуклеотидных оснований; онкоген myc появляется во многих злокачественных новообразованиях в результате амплификации.
В последующем были изучены механизмы, посредством которых большинство, если не все онкогены вызывают трансформацию клеток. Это стало возможным благодаря определению путей, посредством которых клетки регулируют свой собственный рост. Рост иделение нормальной клетки вткани контролируется преимущественно ее окружением. В норме клетка редко или даже никогда не определяет скорости своего деления, а только реагирует на сигналы от окружающих клеток.
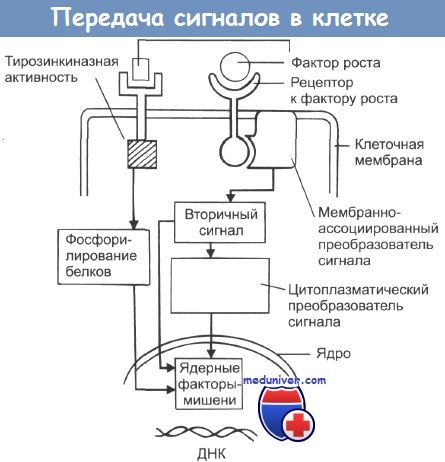
Эти сигналы, стимулирующие или подавляющие рост, передаются посредством ростовых факторов, выделяемых окружающими клетками. Ростовые факторы попадают в межклеточное пространство и связываются с рецепторами на поверхности клеток-мишеней. Клетки реагируют на сигналы ростовых факторов активацией механизмов синтеза клеточных структур, удвоением ДНК и делением. Нормальная клетка никогда не запускает программу роста, если она не получила внешнего сигнала. Каждая клетка обладает сложной системой, позволяющей ей получать ростовые сигналы, обрабатывать их и запускать программу деления. Эта система состоит из большого количества белков, ответственных за получение ростовых сигналов и передачу их в клетку. К этим белкам относятся:
1) рецепторы на клеточной поверхности, распознающие наличие во внеклеточном пространстве ростовых факторов и передающие сигнал во внутриклеточное пространство;
2) белки внутриклеточной сигнальной системы, которые активируются поверхностными рецепторами и затем передают сигнал дальше в клетку;
3) ядерные транскрипционные факторы, активирующиеся в ответ на сигнал, переданный белками внутриклеточной сигнальной системы, и, в свою очередь, активирующие широкий спектр клеточных генов.
Активирующиеся гены руководят программой роста клетки; именно эти гены определяют те события, которые вместе взятые приведут к делению клетки. Протоонкогены кодируют многие белки в этой сложной сигнальной системе, позволяющей нормальной клетке отвечать на экзогенные ростовые факторы. Белки онкогенов участвуют в сигнальной системе, отбирая ее неправильно функционирующие версии нормальных компонентов, и вызывают ее постоянную стимуляцию в отсутствие внешних ростовых сигналов. В результате клетка постоянно растет, даже если окружающая среда не содержит каких-либо факторов, в норме необходимых для клеточного роста.
Некоторые исследователи указывают, что в злокачественной клетке должны присутствовать как минимум две мутации в протоонкогенах, причем только строго определенные мутации могут вызывать злокачественную трансформацию. Это связано с тем, что отдельные онкогены, даже если они служат важными регуляторами клеточного метаболизма, сами по себе не способны индуцировать злокачественное новообразование. Данная точка зрения подтверждается выявлением в опухолевых клетках более 10 различных онкогенов. Однако при тщательном исследовании ожидаемые нарушения обнаруживают лишь примерно в 20 % опухолей.
Ни одна опухоль не несет ни одной пары сочетанных нарушений из выявляемых в культивируемых линиях злокачественных клеток. Предполагается также, что врожденные мутации, ответственные за предрасположенность людей к раку, не есть онкогены. Это объясняется существованием рецессивных антионкогенов, получивших название генов-супрессоров, тоже играющих крайне важную роль в развитии опухолей.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Лучшие виниры подробности на сайте.
Онкогены и протоонкогены как причины рака
• Онкогены способствуют росту и делению клеток
• Супрессоры опухолей подавляют рост и деление клеток
• В геноме клеток присутствует много протоонкогенов
• Опухолеродные вирусы несут онкогены
• В результате генетических изменений протоонкогены могут превратиться в мощные онкогены
Действие на клетку мутагена может вызвать повреждение генов, являющихся положительными или отрицательными регуляторами роста и пролиферации клеток. Для обеспечения хорошо сбалансированного функционирования контрольной системы обычно, в противоположных направлениях, действуют два типа регуляторных генов. Положительные регуляторы, которые обеспечивают нормальные процессы роста и деления клеток, называются протоонкогенами.
После мутации эти гены активируются, превращаясь в онкогены. Отрицательные регуляторы, которые обычно функционируют, ограничивая пролиферацию, называются супрессорами опухоли. Эти гены становятся участниками процесса канцерогенеза, когда они инактивируются за счет мутации, лишая клетку способности подавлять опухолевый рост.
Чтобы понять, каким образом исследования ретровирусов привели к открытию онкогенов, давайте совершим краткий экскурс в историю. Онкогены были открыты раньше, чем опухолевые супрессоры. Начало этих исследований восходит к работе Пейтона Рауса, который в 1910 г. описал вирус, выделенный из опухоли соединительной ткани (саркомы) крыла цыпленка, которого принес к нему в лабораторию на Лонг-Айленде местный фермер.
Когда Раус измельчил саркому и профильтровал тканевой экстракт, он обнаружил вещество, которое проходило через фильтр и при последующем введении здоровому цыпленку индуцировало у него развитие саркомы соединительной ткани. Раус повторил эксперимент, введя профильтрованный экстракт от второго цыпленка третьему, у которого вскоре также на месте инъекции развилась саркома. Поскольку агент, вызывающий образование опухоли, проходил через фильтр, то, по определению, он соответствовал вирусу, а не бактериям (которые, по размерам будучи гораздо крупнее, должны были задерживаться фильтром).
Гораздо позже выяснилось, что обнаруженный Раусом вирус, который стали называть вирусом саркомы Рауса (RSV), сильно отличается от большинства других вирусов, которые попадают в клетку, размножаются там и затем вызывают ее гибель с высвобождением вирусных частиц, инфицирующих соседние клетки. RSV, наоборот, сохранял жизнеспособность инфицированной клетки. Инфицированная клетка быстро приобретала многие черты, свойственные раковым, включая способность к росту в суспензии, измененяла форму и приобретала способность образовывать опухоль (т. е. превращалась в злокачественную); иными словами она становилась трансформированной.
Более того, когда инфицированные и трансформированные клетки росли и делились, у них в потомстве продолжали проявляться признаки рака. В этих клетках присутствовал геном RSV, благодаря чему у них поддерживался злокачественный рост. Фактически злокачественный рост становится наследуемым признаком, передающимся от клетки к потомству, и для проявления которого необходимо постоянное присутствие генома RSV.
Анализ RSV, проведенный в начале 1970-х гг., показал, что он представляет собой ретровирус, содержащий молекулу одноцепочечной РНК, небольшого размера. Было продемонстрировано, что за наступление всех канцерогенных изменений под действием RSV отвечает один ген, обозначенный src, который поэтому стал считаться онкогеном. Для ряда исследователей оказались крайне неожиданными далеко идущие последствия деятельности этого гена, поскольку они свидетельствовали о том, что ген может проявлять плейотропное действие, т. е. способен одновременно индуцировать в клетке множество изменений. Такой спектр изменений позволяет клеткам, трансформированным под действием RSV, размножаться в тканях цыпленка, что в конце концов приводит к развитию крупной опухоли.
К своему удивлению, в 1975 г. исследователи обнаружили, что в здоровых клетках также присутствует обычный вариант гена src, который, очевидно, играет существенную роль в развитии здоровых клеток и организма. Такая обычная версия вируса была названа протоонкоген, поскольку она могла служить предшественником онкогена, переносимого RSV.
Оказалось, что src ген стал частью генома RSV после того, как предок ретровируса, с отсутствующим собственным src геном, инфицировал клетку цыпленка, сделал копию клеточного src гена (иногда называемого c-src) и включил ее в свой геном. После приобретения src гена гибридный вирус превратил его в онкоген (v-src). После этого получившийся опухолевый вирус — теперь RSV — мог трансформировать инфицированные клетки, переводя их в опухолевые. На рисунке ниже показаны структуры белков v-Src и c-Src.
Было также показано, что ряд других онкогенных ретровирусов обладают приобретенными и измененными нормальными клеточными генами, подобно тому как это имеет место в случае RSV. Вирус птичьего миелобластоза захватил ген myc; вирус крысиной саркомы Харви несет H-ras, а вирус кошачьей саркомы несет онкоген fes. В каждом случае эти онкогены, связанные с вирусом, образовались из предсуществующего нормального клеточного протоонкогена.
Следовательно, в геноме животных содержится довольно много этих протоонкогенов, существование которых подтверждается наличием генома того или иного трансформирующего ретровируса.
Другая группа онковирусов вызывает трансформацию клеток по совершенно другому механизму. Для этих вирусов характерен геном, состоящий из двухцепочечной ДНК, и к ним относится вирус папилломы, вызывающий образование бородавок и рак кожи у кроликов, а также карциному шеи у человека. К этой же группе принадлежит вирус SV40 и полиомы, которые вызывают различные опухоли у грызунов, а также вирусы, имеющие отношение к вирусу герпеса, такие как вирус Эпштейна-Барра (EBV), который является причиной развития лимфом у африканского населения и карцином носоглотки в странах Юго-Восточной Азии. ДНК-содержащие онкогенные вирусы продуцируют белки, индуцирующие опухоли (онкобелки), которые не имеют ничего общего с обычными белками, регулирующими рост здоровых клеток.
Напротив, онкобелки связываются с ними и нарушают их функции. Например, SV40, аденовирус и вирус папилломы продуцируют белки, которые связываются и инактивируют белки-супрессоры опухоли. При этом они создают в клетке примерно такое же состояние, которое наблюдается, когда клетка теряет функцию генов-супрессоров опухолей при их инактивации в результате мутации. Рисунок ниже иллюстрирует, каким образом вирусные онкобелки связываются с клеточными белками.
Работы по онкогенным вирусам позволили сделать три вывода, которые способствовали нашему пониманию молекулярных основ происхождения опухолей. Во-первых, клеточный геном содержит многочисленные протоонкогены. Во-вторых, в результате генетических изменений, по крайней мере вызванных ретровирусами, эти протоонкогены способны превращаться в активированные онкогены. В-третьих, каждый из этих генов, превратившись в активированный онкоген, может оказывать плейотропное действие, формируя ряд особенностей раковых клеток.
Вместе с тем, исследования ДНК-содержащих онкогенных вирусов помогли понять молекулярные механизмы действия генов-супрессоров опухолей.

Сравнение первичных структур вирусного и клеточного белка Src показывает,
что они различаются несколькими аминокислотами.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Представления о молекулярно-клеточных механизмах онкогенной трансформации клеток претерпели значительную эволюцию на протяжении XX века и до настоящего времени [18, 20, 25, 32, 34].
Как указывалось выше, инициирующими этиологическими факторами малигнизации клетки являются разнообразные по природе группы канцерогенов химической, физической, биологической природы, в том числе вирусы, гормоны и генотоксические продукты их метаболизма [13, 26, 63].
Естественно, что при чрезвычайной гетерогенности этиологических факторов неоплазий не могла сформироваться достаточно быстро доминирующая концепция механизмов развития онкогенной трансформации клеток, их активации или промоции опухолевого роста с последующей опухолевой прогрессией. В ранних концепциях канцерогенеза делали акцент на эпигеномных механизмах развития неоплазий, и, безусловно, ряд положений этого направления носит не только исторический характер, но может быть в определенной степени ассоциирован с современными вирусо-генетической и онкогенной теориями канцерогенеза.
Согласно данным ряда исследователей, первичное изменение свойств цитоплазматической мембраны под влиянием канцерогенных углеводородов, онкогенных вирусов является одним из пусковых механизмов последующего изменения генетического аппарата и нарушений регуляции их митотического цикла [108]. Эта концепция канцерогенеза была актуальна в период обнаружения отсутствия контактного ингибирования опухолевых клеток в монослойной культуре.
Как оказалось далее, в механизмах контактного ингибирования клеток важная роль отводится активации мембранной аденилциклазы и увеличению уровня цАМФ, тормозящего митотическую активность клеток. Понижение концентрации цАМФ в мембранах клеток под влиянием различных канцерогенов ведет к неконтролируемой митотической активности. Эта точка зрения имела определенную значимость в понимании пусковых механизмов канцерогенеза, поскольку для многих гормонов, регулирующих метаболизм клеток, их митотическую активность, характерен преимущественно мембранный тип рецепции (АКТГ, СТГ, инсулин, пролактин и др.).
Практически одновременно с мембранной концепцией канцерогенеза создавались митохондриальная и лизосомальная теории развития неоплазий, согласно которым актомиозиновый белок митохондриальных мембран оказывается аномальным у малигнизированных клеток и утрачивает чувствительность к регулирующим влияниям АТФ; при этом гликолитическая реакция опухолевой клетки стимулируется митохондриальными факторами, поступающими постоянно в гиалоплазму, а возрастание концентрации АТФ не подавляет этот процесс.
Одним из классических признаков неоплазий является нарушение регуляции дифференцировки и митотической активности клеток, в связи с чем указанная проблема затрагивается в той или иной форме в разных концепциях [1]. Однако до настоящего времени одной из ведущих концепций канцерогенеза является мутационная теория, согласно которой все канцерогены обладают мутагенной активностью, хотя не все мутагены являются канцерогенами.
Практически все изученные канцерогены индуцируют разрывы фосфодиэстеразных связей в молекуле ДНК. Вначале канцерогены интенсивно связываются с ДНК чувствительных клеток. Обнаружена прямая корреляция между чувствительностью животных и их органов к малигнизирующему действию веществ и степенью их связывания с ДНК [42].
В последующие годы важная роль в развитии онкогенной трансформации клеток и опухолевой прогрессии отведена свободным радикалам. Учитывая значимость индукции избыточных концентраций свободных радикалов в канцерогенезе, необходимо прежде всего остановиться на активации процессов липопероксидации, инициируемой активными формами кислорода (АФК) и в то же время являющейся источником образования значительного количества вторичных эндогенных свободных радикалов [7, 8].
Как известно, активные формы кислорода вступают во взаимодействие с полиненасыщенными жирными кислотами (ПНЖК): линолиевой, линоленовой, арахидоновой – важнейшими компонентами фосфолипидов биологических мембран. Отрыв водорода от молекулы ПНЖК при участии АФК приводит к перемещению двойных связей с образованием гидроперекисей диеновых коньюгатов, которые затем метаболизируются во вторичные (малоновый диальдегид) и третичные продукты липопероксидации [66]. Перекисное окисление липидов затрагивает прежде всего фосфолипиды цитоплазматических мембран клеток, нарушая при этом энергозависимый трансмембранный перенос субстратов, процессы межклеточного взаимодействия. Биологическая активность АФК связана с синтезом простагландинов, лейкотриенов окислительной модификацией белков, нуклеиновых кислот, липидов. Одним из проявлений окислительной модификации белка является инактивация около 240 ферментов, в частности, СОД, ацетил-КоА-гидролазы, каталазы, миелопероксидазы, цитохрома Р450 [22, 66].
Дезинтеграция белка в основном возникает под влиянием гидроксильного радикала, образующегося в организме в процессе реакции взаимодействия супероксида и перекиси водорода с металлами переменной валентности. Объектами окисления в молекуле ДНК под влиянием гидроксильного радикала являются углеводные компоненты, фосфатные группировки, азотистые основания. Наиболее чувствительным к окислительной деструкции азотистым основанием является гуанин, модифицированные формы которого составляют 45 % от общего количества окисленных оснований [83, 95].
Установлено, что чувствительность к фрагментации сахарно-фосфатного остатка ДНК под влиянием АФК оказалось более высокой, чем полипептидного остова белково-пептидных субстанций. Гидроксильный радикал, действуя на ДНК, может отрывать атом водорода от дезоксирибозофосфата, что ведет к его расщеплению и освобождению азотистых оснований. При этом образуются высокотоксичные производные альдегиды.
Данные, опубликованные в последние годы, убедительно свидетельствуют о том, что активные формы кислорода, оксид азота и его производные в сочетании с инфекционными патогенными факторами, бактериями и вирусами, являются ключевыми факторами канцерогенеза [2, 35, 36].
Детальный обзор литературы по этому вопросу приведен в работе Х. Маеда, Т. Акаике (1998). Кислородные радикалы, а также оксид азота могут повреждать ДНК, вызывая мутацию. Мутагенный и канцерогенный эффекты указанных соединений резко возрастают при одномоментной, избыточной продукции, сопровождающейся их взаимодействием с образованием пероксинитрита. Последний участвует в различных внутриклеточных метаболических процессах: нитровании остатков тирозина в белках, подавлении транспорта электронов в митохондриях, в окислении тиоловых соединений. Пероксинитрит является ДНК-расщепляющим агентом. Вышеуказанные химические реакции с участием пероксинитрита могут инициировать апоптоз, мутации, онкогенную трансформацию клеток.
Как указывалось выше, в механизмах индукции канцерогенеза важная роль отводится онкогенным ДНК- и РНК-содержащим вирусам, способным инкорпорировать свою ДНК или ДНК-копию в геном хозяина с последующей возможной онкогенной трансформацией клетки в случае экспрессии протоонкогенов.
Установлено, что РНК-содержащие онкогенные вирусы являются членами семейства ретровирусов, характеризуются наличием липидной оболочки и двух односпиральных РНК, фермента РНК-зависимой ДНК-полимеразы, необходимой для репродукции вируса. Наличие этого фермента обеспечивает обратную транскрипцию вирусной РНК- в ДНК-копию, интегрирующую с геномом клетки [71].
Группа РНК-содержащих вирусов включает следующие разновидности: непатогенную для человека группу вирусов (род А); медленно трансформирующийся вирус гормонзависимой карциномы молочной железы морских свинок и, возможно, человека (род В); дефектные быстро трансформирующиеся и недефектные медленно трансформирующиеся вирусы (род С); род Д – включает вирусы приматов и вирус перевиваемых раковых клеток человека.
ДНК-содержащие онкогенные вирусы подразделяются на следующие семейства:
1. Семейство Poxviridae, содержит, в частности, вирус контагиозного моллюска человека.
2. Семейство Herpes viridae, к которому относится вирус Эпштейн-Барра человеа, вызывающий лимфому Беркитта, цитомегаловирус человека – тип 5.
3. Семейство Adenoviridae – представителями которого являются аденовирусы человека.
4. Семейство Papovaviridae, представителями которого являются вирусы папилломы крыс, хомяков, обезьян, человека.
ДНК-содержащие вирусы внедряют свою ДНК в геном хозяина при участии ферментов эндонуклеаз и липаз, а за счет наличия генов – промоторов – вирусы инициируют транскрипцию генов, следующих за ДНК-вирусами. Последствия внедрения ДНК-вирусов в геном хозяина зависят от зоны инкорнации: интронов, экзонов, протоонкогенов, антионкогенов. Если ДНК-содержащие вирусы встраивают в геном хозяина клетки регуляторы экспрессии протоонкогенов, возможна малигнизация клетки [54].
Механизмы онкогенной трансформации клеток под влиянием ДНК-содержащих вирусов могут быть весьма разнообразны: за счет индукции ранних онкобелков, так называемых Т-антигенов, усиления экспрессии рецепторов экзогенных ростовых факторов. Большие и средние Т-белки ряда ДНК-содержащих вирусов выключают контактное ингибирование пролиферации клеток, препятствуют действию антионкогена р53.
Как известно, вирусо-генетическая теория Л.А. Зильбера явилась основной для формирования современной онкогенной теории канцерогенеза. На смену вирусогенетической теории канцерогенеза пришли теории онкогенов, протоонкогенов и антионкогенов [30, 31, 65, 120].
В настоящее время, очевидно, что в опухолевой трансформации клеток, возникающей под влиянием различных индукторов канцерогенеза, принципиально участвуют следующие категории генов:
1. Онкогены- стимуляторы функций.
2. Гены роста и пролиферации клеток (Myc, Ras, Los, ABL и другие).
3. Антионкогены (потеря функции).
4. Гены, отвечающие за программированную смерть клетки (апоптоз):
– отменяющие программированную смерть: Bcl-2 (стимуляция функций);
– гены смерти клеток – р53 (потеря функции).
Онкогены как специфический химический материал, кодирующий информацию об определенном химическом продукте, впервые были идентифицированы в составе ретровирусов. Геном типичного не трансформирующего ретровируса представляет собой две молекулы односпиральной РНК. Основные гены вируса относятся к трем регионам: gag кодирует структурные белки вирион частицы, env– белки оболочки вириона, ген pol – несет информацию об обратной транскрипции. Последний обеспечивает образование ДНК- копии на матрице РНК-вируса.
Согласно гипотезе онкогенов, гены ретровирусов, попавшие в геном человека в процессе эволюции, переходят по наследству в ряде поколений, проявляют себя в раннем онтогенезе, а затем подавляются внутриклеточными репрессорами. С возрастом под влиянием различных канцерогенов физической, химической, биологической природы возникают экспрессия вирусных онкогенов и усиление продукции ими онкобелков, ответственных за малигнизацию клетки. Онкогенные свойства нетрансформирующих ретровирусов обусловлены наличием в их геноме V-онкогенов, причем большинство из 50 V-онкогенов имеют клеточные прототипы – С-протоонкогены.
Высказывается мысль, что ретровирусы не только могут вносить в определенные позиции клеточного генома V-онкогены, но и способны быть промоторами для усиленной экспрессии протоонкогенов клеток. Считается, что в ходе совместной эволюции ретровирусов и клеток происходят захват клеточных протонкогенов вирусами и их перенос [24].
Развитие теории онкогенов нашло отражение в концепции Темина (1972) о протовирусах, протоонкогенах, согласно которой предсуществующий аналог вируса не является результатом инфекции, а нормальным клеточным геном, необходимым для роста и онтогенеза клеток, причем нормальные клетки не содержат вирусных онкогенов, но зависят от контролируемой экспрессии их клеточных аналогов.
В механизмах развития неоплазий онкогенные ретровирусы играют неоднозначную роль: различают быстро- и медленно-трансформирующие вирусы. Быстротрансформирующие вирусы дефектны по структуре, утратили часть своих поздних репликативных генов и приобрели взамен видоизмененные клеточные гены-V-онкогены, которые и вызывают неопластическую трансформацию при повторной интеграции в клеточный геном. Для полного цикла репликации этим вирусом требуются вирусы-помощники. Клеточные протоонкогены являются прототипами V-онкогенов, консервативными регуляторами клеточной дифференцировки.
Встраивание быстро-трансформирующего реторовируса может либо привести к экспрессии в клетке V-онкогена, либо вирусные промоторы и энхансеры встраиваются рядом с протоонкогенами клетки, вызывая их экспрессию.
Таким образом, встраивание ретровирусов в геном клетки приводит к гиперэкспрессии протоонкогенов, переход их в онкогены с последующей малигнизацией клетки [20, 23, 30, 64].
Что касается механизмов индукции неоплазий химическими канцерогенами с точки зрения современных теорий канцерогенеза – протоонкогенов, онкогенов, антионкогенов, то необходимо остановиться на анализе лишь некоторых работ, посвященных данной проблеме.
Как известно, химические канцерогены, подобно биологическим, способны вызывать развитие мутаций и активацию протоонкогенов [25, 64]. Под влиянием химических канцерогенов возможна онкогенная трансформация в процессе амплификации ДНК. Установлено, что амплификация гена резистентности на фоне воздействия цитостатиков нередко возникает при раке кишечника и является причиной устойчивости неоплазий к химиотерапии. При ряде онкологических заболеваний желудочно-кишечного тракта возникает амплификация онкогенов erbB2, mys, SRS. Индукция развития опухолей нитрозмочевиной связана с амплификацией и активацией N-ras; в опухолях, индуцированных гамма-облучением, активен Ras-H. В ходе химического канцерогенеза отмечено гипометилирование протоонкогена Ras-H, приводящего к развитию генной мутации.
В опухолях, индуцированных химическими канцерогенами, отмечены транскрипции ряда других онкогенов (c-ras и c-mys), связанные с гипометилированием протоонкогена либо его амплификацией. В ходе химического канцерогенеза нарушается зависимость экспрессии c-mys (но не c-ras) от клеточного цикла. Таким образом, многие химические соединения или физические воздействия, а также вирусы могут вызывать мутации ДНК, не летальные для клеток и провоцирующие экспрессию протоонкогенов или депрессию антипротоонкогенов [108]. Последнее приводит к трансформации нормальной клетки в опухолевую.
Эпигенетический механизм канцерогенеза связан с нарушением регуляции клеточного роста, функции клетки и экспрессии генов без повреждения генома. При эпигенетическом канцерогенном эффекте эндогенных или экзогенных канцерогенных факторов возникает инактивация белков-продуктов антипротоонкогенов или активация пострецепторных передатчиков ростовых факторов. Такое воздействие, как правило, не вызывает неоплазии, но усиливает ростовые эффекты, способствует пролиферации мутантного клона и формированию распознаваемой неоплазии. Эффект канцерогенов-мутагенов называют инициирующим, а коканцерогенов – активирующим.
Таким образом, в настоящее время очевидны следующие механизмы активации протоонкогенов:
1) амплификация протоонкогенов, в результате чего резко возрастает их общая активность, что может привести к малигнизации клетки;
2) мутации протоонкогенов, приводящие к их активации, и ингибиция антипротоонкогенов;
3) транслокация протоонкогенов в локус с функционирующим промотором;
4) аддукция промотора рядом с протоонкогеном. В качестве промотора могут выступать ДНК-копии определенных участков онкорнавирусов, а также мобильные генетические структуры, способные перемещаться и встраиваться в различные участки генома.
В геноме человека предполагается наличие около 100 протоонкогенов, выполняющих следующие функции:
1) кодирование ростовых факторов, их рецепторов и пострецепторных передатчиков;
2) кодирование блокаторов запрограммированной гибели клеток, контактного ингибирования пролиферации.
Трансформация протоонкогенов в онкогены приводит к их экспрессии и синтезу онкобелков. При этом онкобелки продуцируются перманентно в увеличенном количестве или в качественно измененном состоянии.
Ниже представлены несколько групп протоонкогенов, антионкогены, и кодируемые ими белки [30, 31, 32].
Читайте также: