
Малярия при серповидноклеточной анемии
Обновлено: 25.04.2024
Генетика серповидноклеточной анемии. Наследование
HbS был первым обнаруженным аномальным гемоглобином с высоким клиническим значением. Он возникает вследствие замены единственного нуклеотида, которая изменяет кодон шестой аминокислоты В-глобина глутаминовой кислоты на валин (GAG -> GTG: Glu6Val).
Гомозиготность по данной мутации — причина серповидноклеточной анемии, серьезного заболевания, часто встречающегося в некоторых частях света. Болезнь имеет характерное географическое распределение, чаще всего встречается в Экваториальной Африке и реже всего в Средиземноморье, Индии и странах, в которые мигрировали люди из этих регионов. С этой, обычно фатальной в раннем детстве болезнью рождаются около 1 из 600 афроамериканцев, хотя все более частым становится более долгое выживание.
Случаи, когда бы это могло происходить in vivo, редки, хотя гетерозиготы имеют риск инфаркта селезенки, особенно при полетах на большой высоте в самолетах с низким давлением в кабине. Гетерозиготное состояние наблюдают приблизительно у 8% афроамериканцев, но в областях, где частота гена высокая (например, в Западной Центральной Африке), вплоть до 25% новорожденных — гетерозиготы.
Молекулярная патология HbS - серповидноклеточной анемии
Около 50 лет тому назад Ингрэм обнаружил, что аномалия HbS связана с заменой одной из 146 аминокислот в В-цепи молекулы гемоглобина. Все клинические проявления наличия HbS — последствия этого единственного изменения в гене В-глобина. Это было первой демонстрацией того, что мутация в структурном гене может вызывать замену аминокислоты в соответствующем белке. Поскольку аномалия HbS локализуется в В-цепи, формула HbS может быть записана как а2b2s или, более точно, а2Ab2s.
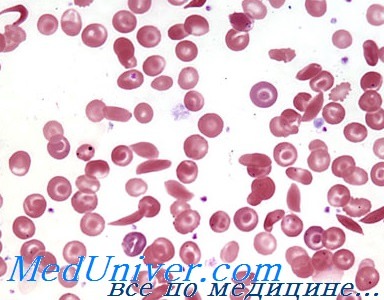
Гетерозиготы имеют смесь двух типов гемоглобинов (НbА и HbS), обозначаемых а2Аb2А, а2Аbs, а также гибридный тетрамер гемоглобина, обозначаемый как a2AbA,bs.
Серповидноклеточность и ее последствия
Молекулы гемоглобина, содержащие мутантные субъединицы b-глобина, нормальны по их способности выполнять их главную функцию связывания кислорода (если они не полимеризованы, как указано далее), но в ненасыщенной кислородом крови они растворимы в пять раз меньше по сравнению с нормальным гемоглобином. Относительная нерастворимость дезоксигемоглобина S является физической основой феномена серповидноклеточности.
В условиях низкой кислородной напряженности молекулы HbS собираются в форме полимеров, формирующих стержни или волокна, искажающие форму эритроцитов. Эти уродливые эритроциты деформируются хуже, чем в норме, и, в отличие от нормальных красных кровяных клеток, не могут сжиматься, проходя через капилляры, тем самым блокируя ток крови и вызывая локальную ишемию.
Происхождение мутаций гемоглобина S
У большинства лиц африканского происхождения нормальный ген b-глобина содержится в пределах фрагмента рестрикции размером в 7,6 килобазы ДНК. В то же время в определенных частях Африки, например в Гане и почти у 70% афроамериканцев, аллель серповидноклеточного глобина часто обнаруживают во фрагменте размером в 13 килобаз. Частая ассоциация серповидноклеточного глобина с 13-килобазовым фрагментом — поразительный пример неравновесного сцепления.
В других частях Африки (например, в Кении) мутация серповидноклеточности обычно связана с фрагментом размером в 7,6 килобазы. Эти находки позволяют утверждать, что мутация серповидноклеточности возникла в Западной Африке в хромосоме, которая содержала ген р-глобина во фрагменте длиной 13 килобаз, и что подобная мутация, по крайней мере, один раз, независимо произошла где-то еще. Защита от малярии, обеспечиваемая данной мутацией у гетерозигот, обеспечила ее высокую частоту в областях, пораженных малярией.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Серповидно-клеточная анемия – наследственная гемоглобинопатия, обусловленная синтезом аномального гемоглобина S, изменением формы и свойств эритроцитов крови. Серповидно-клеточная анемия проявляется гемолитическими, апластическими, секвестрационными кризами, тромбозами сосудов, костно-суставными болями и припухлостью конечностей, изменениями скелета, сплено- и гепатомегалией. Диагноз подтверждается по данным исследования периферической крови и пунктата костного мозга. Лечение серповидно-клеточной анемии является симптоматическим, направленным на предупреждение и купирование кризов; может быть показано переливание эритроцитов, прием антикоагулянтов, проведение спленэктомии.



Общие сведения
Серповидно-клеточная анемия (S-гемоглобинопатия) – разновидность наследственной гемолитической анемии, характеризующаяся нарушением структуры гемоглобина и присутствием в крови эритроцитов серповидной формы. Заболеваемость серповидно-клеточной анемией распространена, главным образом, в странах Африки, Ближнего и Среднего Востока, Средиземноморского бассейна, Индии. Здесь частота носительства гемоглобина S среди коренного населения может достигать 40%. Любопытно, что больные серповидно-клеточной анемией имеют повышенную врожденную устойчивость к заражению малярией, поскольку малярийный плазмодий не может проникнуть в эритроциты серповидной формы.
Причины
В основе серповидно-клеточной анемии лежит генная мутация, обусловливающая синтез аномального гемоглобина S (HbS). Дефект структуры гемоглобина характеризуется заменой глутаминовой кислоты валином в ß-полипептидной цепи. Образующийся при этом гемоглобин S после потери присоединенного кислорода приобретает консистенцию высокополимерного геля и становится в 100 раз менее растворимым, чем нормальный гемоглобин А. В результате этого эритроциты, несущие деоксигемоглобин S, деформируются и приобретают характерную полулунную (серповидную) форму. Измененные эритроциты становятся ригидными, малопластичными, могут закупоривать капилляры, вызывая ишемию тканей, легко подвергаются аутогемолизу.
Наследование серповидно-клеточной анемии происходит по аутосомно-рецессивному типу. При этом, гетерозиготы наследуют дефектный ген серповидно-клеточной анемии от одного из родителей, поэтому, наряду с измененными эритроцитами и HbS, имеют в крови и нормальные эритроциты с HbА. У гетерозиготных носителей гена серповидно-клеточной анемии признаки заболевания возникают лишь в определенных условиях. Гомозиготы наследуют по одному дефектному гену от матери и от отца, поэтому в их крови присутствуют только серповидные эритроциты с гемоглобином S; заболевание развивается рано и протекает тяжело.
Таким образом, в зависимости от генотипа, в гематологии различают гетерозиготную (HbAS) и гомозиготную (HbSS, дрепаноцитоз) форму серповидно-клеточной анемии. К редко встречающимся вариантам заболевания относятся промежуточные формы серповидно-клеточной анемии. Обычно они развиваются у двойных гетерозигот, несущих один ген серповидно-клеточной анемии и другой дефектный ген - гемоглобина C (HbSC), серповидной β-плюс (HbS/β +) или β-0 (HbS/β0) талассемии.
Симптомы серповидно-клеточной анемии
Гомозиготная серповидно-клеточная анемия обычно проявляется у детей к 4-5 месяцу жизни, когда увеличивается количество HbS, а процентное содержание серповидных эритроцитов достигает 90%. В таких случаях раннее возникновение гемолитической анемии у ребенка обуславливает задержку физического и умственного развития. Характерны нарушения развития скелета: башенный череп, утолщение лобных швов черепа в виде гребня, кифоз грудного или лордоз поясничного отдела позвоночника.
В развитии серповидно-клеточной анемии выделяют три периода: I - с 6 месяцев до 2-3 лет, II - с 3 до 10 лет, III - старше 10 лет. Ранними сигналами серповидно-клеточной анемии служат артралгии, симметричное опухание суставов конечностей, боли в груди, животе и спине, желтушность кожи, спленомегалия. Дети с серповидно-клеточной анемией относятся к категории часто болеющих. Степень тяжести течения серповидно-клеточной анемии тесно коррелирует с концентрацией HbS в эритроцитах: чем она выше, тем тяжелее выражена симптоматика.
В условиях интеркуррентной инфекции, стрессовых факторов, обезвоживания, гипоксии, беременности и пр. у больных данным видом наследственной анемии могут развиваться серповидно-клеточные кризы: гемолитический, апластический, сосудисто-окклюзионный, секвестрационный и др.
При развитии гемолитического криза состояние больного резко ухудшается: возникает фебрильная лихорадка, в крови повышается непрямой билирубин, усиливается желтушность и бледность кожных покровов, появляется гематурия. Стремительный распад эритроцитов может привести к анемической коме. Апластические кризы при серповидно-клеточной анемии характеризуются угнетением эритроидного ростка костного мозга, ретикулоцитопенией, снижением гемоглобина.
Следствием депонированием крови в селезенке и печени служат секвестрационные кризы. Они сопровождаются гепато- и спленомегалией, сильными болями в животе, резкой артериальной гипотонией. Сосудисто-окклюзионные кризы протекают с развитием тромбоза сосудов почек, ишемии миокарда, инфаркта селезенки и легких, ишемического приапизма, окклюзии вен сетчатки, тромбоза мезентериальных сосудов и др.
Гетерозиготные носители гена серповидно-клеточной анемии в обычных условиях ощущают себя практически здоровыми. Морфологически измененные эритроциты и анемия у них возникают только в ситуациях, связанных с гипоксией (при тяжелой физической нагрузке, авиаперелетах, восхождении в горы и др.). Вместе с тем, остро развившийся гемолитический криз при гетерозиготной форме серповидно-клеточной анемии может иметь летальный исход.
Осложнения
Хроническое течение серповидно-клеточной анемии с повторными кризами приводит к развитию целого ряда необратимых изменений, нередко становящихся причиной гибели больных. Примерно у трети больных отмечается аутоспленэктомия – сморщивание и уменьшение размеров селезенки, вызванное замещением функциональной ткани рубцовой. Это сопровождается изменением иммунного статуса больных серповидно-клеточной анемией, более частым возникновением инфекций (пневмонии, менингита, сепсиса и др.).
Исходом сосудисто-окклюзионных кризов могут стать ишемические инсульты у детей, субарахноидальные кровоизлияния у взрослых, легочная гипертензия, ретинопатия, импотенция, почечная недостаточность. У женщин с серповидно-клеточной анемией отмечается позднее становление менструального цикла, склонность к самопроизвольному прерыванию беременности и преждевременным родам. Следствием ишемии миокарда и гемосидероза сердца служит возникновение хронической сердечной недостаточности; повреждения почек - хронической почечной недостаточности.
Длительный гемолиз, сопровождаемый избыточным образованием билирубина, приводит к развитию холецистита и желчнокаменной болезни. У больных серповидно-клеточной анемией часто возникают асептические некрозы костей, остеомиелит, язвы голеней.
Диагностика
Диагноз серповидно-клеточной анемии выставляется гематологом на основании характерных клинических симптомов, гематологических изменений, семейно-генетического исследования. Факт наследования ребенком серповидно-клеточной анемии может быть подтвержден еще на этапе беременности с помощью биопсии ворсин хориона или амниоцентеза.
В периферической крови отмечается нормохромная анемия (1-2х1012/л), снижение гемоглобина (50-80 г/л), ретикулоцитоз (до 30%). В мазке крови обнаруживаются серповидно измененные эритроциты, клетки с тельцами Жолли и кольцами Кабо. Электрофорез гемоглобина позволяет определить форму серповидно-клеточной анемии – гомо- или гетерозиготную. Изменение биохимических проб крови включает гипербилирубинемию, увеличение содержания сывороточного железа. При исследовании пунктата костного мозга выявляется расширение эритробластического ростка кроветворения.
Дифференциальная диагностика направлена на исключение других гемолитических анемий, вирусного гепатита А, рахита, ревматоидного артрита, туберкулеза костей и суставов, остеомиелита и др.
Лечение серповидно–клеточной анемии
Серповидно-клеточная анемия относится к категории неизлечимых болезней крови. Таким пациентам требуется пожизненное наблюдение гематолога, проведение мероприятий, направленных на предупреждение кризов, а при их развитии – проведение симптоматической терапии.
В период развития серповидно-клеточного криза требуется госпитализация. С целью быстрого купирования острого состояния назначается кислородотерапия, инфузионная дегидратация, введение антибиотиков, обезболивающих средств, антикоагулянтов и дезагрегантов, фолиевой кислоты. При тяжелом течении обострений показано переливание эритроцитарной массы. Проведение спленэктомии не способно повлиять на течение серповидно-клеточной анемии, однако может на время уменьшить проявления заболевания.
Прогноз и профилактика
Прогноз гомозиготной формы серповидно-клеточной анемии неблагоприятный; большая часть пациентов погибает в первое десятилетие жизни от инфекционных или тромбоокклюзионных осложнений. Течение гетерозиготных форм патологии гораздо более обнадеживающее.
Для предупреждения быстро прогрессирующего течения серповидно-клеточной анемии следует избегать провоцирующих условий (обезвоживания, инфекций, перенапряжения и стрессов, экстремальных температур, гипоксии и пр.). Детям, страдающим данной формой гемолитической анемии, в обязательном порядке показана вакцинация против пневмококковой и менингококковой инфекции. При наличии в семье больных серповидно-клеточной анемией необходима медико-генетическая консультация для оценки риска развития заболевания у потомства.
Серповидноклеточная анемия: причины, диагностика, лечение
Этиология и встречаемость серповидноклеточной анемии. Серповидноклеточная анемия (MIM № 603903) — аутосомно-рецессивное заболевание гемоглобина, вызванное миссенс-мутацией гена бета-субъединицы, заменяющей валин на глутаминовую кислоту в 6 положении. Болезнь чаще вызвана гомозиготностью по мутации серповидноклеточности, хотя серповидноклеточную анемию также может вызывать компаундная (составная) гетерозиготность по аллелю серповидноклеточности и аллелям HbC или бета-талассемии.
Распространение серповидноклеточной анемии широко изменяется среди популяций в соответствии с прошлым и настоящим распространением малярии. Мутация серповидноклеточности, как оказалось, несколько повышает сопротивляемость малярии, таким образом, давая преимущество выживания гетерозиготным носителям мутации.
Патогенез серповидноклеточной анемии
Гемоглобин формируется из четырех субъединиц: двух а-субъединиц, кодируемых геном ЯВА в хромосоме 16, и двух бета-субъединиц, кодируемых геном ЯВВ в хромосоме 11. Мутация Glu6Val в бета-субъединице уменьшает растворимость ненасыщенного кислородом гемоглобина и вызывает формирование сети жестких волокнистых полимеров, искажающих строение эритроцита, придавая ему форму серпа. Серповидные эритроциты закупоривают капилляры и вызывают инфаркты.
Первоначально обогащение кислородом заставляет полимер гемоглобина растворяться, и эритроциты восстанавливают нормальную форму; тем не менее, регулярное нарушение формы приводит к необратимому переходу клеток в серповидную форму, впоследствии такие эритроциты удаляются из кровотока в селезенке. Скорость удаления эритроцитов из кровотока превышает возможность их синтеза в костном мозге, что приводит к гемолитической анемии.
Аллельная гетерогенность часто встречается при большинстве менделирующих заболеваний, особенно когда мутантные аллели вызывают снижение функции. Серповидноклеточная анемия — важное исключение из этого правила, поскольку в данном случае единственная специфическая мутация ответственна за уникальные новые свойства HbS. HbC тоже менее растворим, чем HbA, и тоже стремится кристаллизоваться в эритроцитах, уменьшая их деформируемость в капиллярах и вызывая легкий гемолиз, но HbC не формирует полимерные волокна, как HbS. Неудивительно, что другие мутации с новыми функциями, например, мутации в гене FGFR3, вызывающие ахондроплазию, часто имеют аналогичное снижение аллельной гетерогенности, когда фенотип зависит от специфического, уникального изменения функции белка.
Фенотип и развитие серповидноклеточной анемии
Клиническая картина у больных серповидноклеточной анемией обычно проявляется в течение первых двух лет жизни анемией, задержкой развития, спленомегалией, регулярными инфекциями и дактилитами (болезненными припухлостями кистей или стоп, вызванными закупоркой капилляров в небольших костях, обнаруженных у приведенной в примере пациентки).
Инфаркты вследствие закупорки сосудов происходят во многих тканях, вызывая инсульты мозга, острый кардиальный синдром, почечный папиллярный некроз, инфаркты селезенки, язвы ног, приапизм, асептический некроз костей и снижение зрения. Окклюзия сосудов костей вызывает приступы болей, при отсутствии лечения эти болезненные эпизоды могут продолжаться в течение нескольких дней и даже недель. Функциональная аспления вследствие инфарктов и других недостаточно ясных факторов, предрасполагает к бактериальным инфекциям, например, пневомококковому или сальмонеллезному сепсису и остеомиелиту.
Инфекция — основная причина смерти во всех возрастных группах, хотя прогрессирующая почечная и дыхательная недостаточность также нередкие причины смерти на четвертом и пятом десятилетиях жизни. Пациенты также имеют высокий риск развития угрожающей жизни апластической анемии после парвовирусной инфекции, поскольку парвовирусы вызывают временное прекращение образования эритроцитов.
Особенности фенотипических проявлений серповидноклеточной анемии:
• Возраст начала: детство
• Анемия
• Инфаркты
• Аспления
Лечение серповидноклеточной анемии
Конкретному больному серповидноклеточной анемией дать точный прогноз тяжести течения болезни невозможно. Хотя молекулярная основа болезни стала известной раньше других моногенных заболеваний, лечение остается только симптоматическим. Никакой специфический терапии, предохраняющей от процесса образования серповидных эритроцитов, не найдено.
Существенно снижает тяжесть болезни персистенция HbE Исследуется несколько фармакологических препаратов, нацеленных на увеличение концентрации HbF, в этих целях одобрено использование гидрокси-мочевины. Хотя генотерапия имеет шанс улучшить или излечить эту болезнь, эффективная пересадка гена b-глобина не достигнута. Пересадка костного мозга остается единственным доступным в настоящее время лечением, способным помочь при серповидноклеточной анемии.
Из-за 11% смертности, вызванной сепсисом в первые 6 мес жизни, большинство штатов в США проводит неонатальный скрининг на серповидноклеточную анемию с целью проведения профилактики антибиотиками, продолжающейся до 5-летнего возраста.
Риски наследования серповидноклеточной анемии
Поскольку серповидноклеточная анемия — аутосомно-рецессивное заболевание, будущие сибсы больного ребенка имеют 25% риск серповидноклеточной анемии и 50% риск носительства серповидноклеточности. Используя ДНК плода, полученную при БВХ или амниоцентезе, можно провести пренатальную диагностику обнаружением мутации.
Пример серповидноклеточной анемии. Второй раз за полгода семейная пара карибского происхождения обратилась со своей 24-месячной дочерью в отделение неотложной помощи, поскольку девочка не может стоять. В анамнезе отсутствуют повышение температуры, инфекция или травма, и в остальном медицинская история ничем не примечательна; данные предыдущих осмотров соответствовали норме, за исключением низкого уровня гемоглобина и слегка увеличенной селезенки. При текущем осмотре патологии не найдено, за исключением пальпируемого края селезенки и отека стоп.
Стопы болезненны при пальпации, и девочка не хотела вставать на ноги. Оба родителя имели сибсов, умерших в детстве от инфекций, и других сибсов, вероятно, имевших серповидноклеточную анемию. С учетом анамнеза и повторного болезненного увеличения стоп врач проверил ребенка на наличие серповидноклеточной анемии методом электрофореза гемоглобина. Результат этого теста подтвердил наличие HbS.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Сиклемия (дрепаноцитоз, серповидноклеточная анемия, серповидная анемия, гемоглобиноз S) - история изучения, причины
Сиклемия (синонимы: дрепаноцитоз, серповидноклеточная анемия, серповидная анемия, гемоглобиноз S) представляет собой качественное заболевание гемоглобина, при котором основное нарушение состоит в наличии гемоглобина S в эритроцитах. Болезнь характеризуется хронической гемолитической анемией и явлениями сосудистой закупорки, причину которых следует искать в дрепаноцитном преобразовании эритроцитов.
В 1949 г. Neel и Beet доказали наследственный характер болезни, что тяжелая форма отражает гомозиготное состояние, в то время как легкая — гетерозиготное. В том же году Janet Watson высказал мнение о том, что причину заболевания следует искать в наличии иного вида гемоглобина, чем от нормального взрослого человека.
Вскрытие первого аномального гемоглобина сделано Pauling и его сотрудниками, которые, в 1949 г. доказали, что картина электрофоретической миграции гемоглобина страдающих анемией, в крови которых имеются серповидные красные кровяные клетки, иная, чем нормального гемоглобина. Уточнение структурной аномалии гемоглобина S принадлежит Ingram в 1956 г. в связи с внедрением техники „fingerprint".
«Выявление первого аномального гемоглобина — гемоглобина S — составило не только новый этап в развитии знаний о гемоглобинопатиях, но и межевой камень медицинского исследования XX века в связи с обоснованием общей концепции о молекулярных болезнях.
Клинический этап понимания болезни, предшествующий 1949 г., дополнился, в дальнейшем, познанием глубокого патофизиогенетического механизма по последним знаниям о структуре и аномалиях гемоглобина. В этой связи было доказано, что сиклемия — комплексное клиническое заболевание — результат молекулярной аномалии лишь одного белка, замены одной аминокислоты в молекуле гемоглобина, другой.
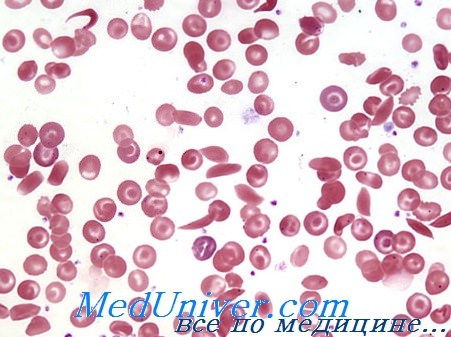
Причины сиклемии - серповидноклеточной анемии
Сиклемия это молекулярное заболевание, причиной которого является генетическая мутация в контролирующих синтез гемоглобина генах.
Эпидемиология сиклемии - серповидноклеточной анемии
Наибольшая частота заболевания наблюдается среди народов экваториальной зоны африканского материка, также на аравийском полуострове, на юге Индии, Цейлоне, Мадагаскаре и среди цветного населения южной и северной Америк. Что касается Европы очаги гемоглобина S описаны в Турции, Греции, южной Италии, в то время как у остальных народов Европы диагноз гемоглобина S редкое явление.
У нас в стране о первых случаях гемоглобина S было сообщено в 1967 г.; в настоящее время на учете коллектива гематологилческого Центра значатся 11 семей с этой генетической аномалией.
Следует отметить, что гемоглобин S создает некоторую устойчивость к вызываемой Plasmodium falciparum малярии. Эта отличительная черта составляет один из факторов, объясняющих высокий показатель частоты сиклемии в данной зоне.
Так, у аномальных индивидов (АА) смертность от малярии высокая; у индивидов с сиклемией (SS) показатель смертности нысокий в связи с собственно тяжестью заболевания; гетерозиготные больные (AS) устойчивы к малярии и продолжительность жизни укладываетвя в норму.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Малярия – трансмиссивная протозойная инфекция, вызываемая патогенными простейшими рода Plasmodium и характеризующаяся приступообразным, рецидивирующим течением. Специфическими симптомами малярии служат повторные приступы лихорадки, гепатоспленомегалия, анемия. В течении лихорадочных приступов у больных малярией четко прослеживаются сменяющие друг друга стадии озноба, жара и пота. Диагноз малярии подтверждается при обнаружении малярийного плазмодия в мазке или толстой капле крови, а также результатами серологической диагностики. Для этиотропной терапии малярии используются специальные противопротозойные препараты (хинин и его аналоги).
Общие сведения
Малярия (перемежающаяся лихорадка, болотная лихорадка) – группа паразитарных заболеваний человека, возбудителями которых выступают различные виды малярийного плазмодия, поражающего преимущественно эритроциты крови и ретикулоэндотелиальную систему. Малярия протекает с лихорадочными пароксизмами, гепатолиенальным и анемическим синдромом. Малярия широко распространена в странах Экваториальной Африки, Юго-Восточной Азии, Океании, Центральной и Южной Америки. Ежегодно в мире регистрируется 350-500 млн. новых инвазий и порядка 1,3-3 млн. летальных исходов от малярии. Высокая заболеваемость малярией в мире объясняется развитием резистентности плазмодиев к специфической терапии, а переносчиков протозойной инфекции – к действию инсектицидов. В связи с увеличением миграционных и туристических потоков завозные случаи малярии все чаще встречаются на территории Европы, в т. ч. в России.
Причины малярии
Малярию вызывают паразитические простейшие, принадлежащие к классу споровиков, роду Plasmodium (малярийные плазмодии). Заболевание человека вызывают 4 вида плазмодиев: P. Vivax (возбудитель трехдневной малярии), P. Malariae (возбудитель четырехдневной малярии), P.falciparum (возбудитель тропической малярии) и P. Ovale (возбудитель овале-малярии, сходной с трехдневной).
Малярийные плазмодии проходят сложный жизненный цикл, включающий бесполое развитие (шизогонию) в организме промежуточного хозяина - человека и половое развитие (спорогонию) в организме главного хозяина - самок комаров Anopheles. Инфицирование комаров происходит при укусах человека, больного малярией или паразитоносителя. При кровососании в желудок комара попадают мужские и женские половые клетки плазмодиев (микро-и макрогаметоциты); здесь происходит их оплодотворение с образованием зиготы, а затем ооцисты. В результате многократного деления ооциста превращается в инвазионные формы плазмодиев - спорозоиты, которые проникают в слюнные железы комара и могут там находиться в течение 2-х месяцев.
Инфицирование человека происходит при укусе инвазированной самкой комара, со слюной которой в кровь промежуточного хозяина проникают спорозоиты. В организме человека возбудитель малярии проходит тканевую и эритроцитарную фазы своего бесполого развития. Тканевая фаза (экзоэритроцитарная шизогония) протекает в гепатоцитах и тканевых макрофагах, где спорозоиты последовательно трансформируются в тканевые трофозоиты, шизонты и мерозоиты. По окончании этой фазы мерозоиты проникают в эритроциты крови, где протекает эритроцитарная фаза шизогонии. В клетках крови мерозоиты превращается в трофозоиты, а затем в шизонты, из которых в результате деления вновь образуются мерозоиты. В конце такого цикла эритроциты разрушаются, а высвободившиеся мерозоиты внедряются в новые эритроциты, где цикл превращений повторяется вновь. В результате 3-4-х эритроцитарных циклов, образуются гаметоциты – незрелые мужские и женские половые клетки, дальнейшее (половое) развитие которых протекает в организме самки комара Anopheles.
Учитывая особенности развития плазмодия, становится очевидным, что основным путем передачи малярии от человека человеку является трансмиссивный, реализуемый посредством укусов самками комара рода Anopheles. Вместе с тем, возможна трансплацентарная передача инфекции во время беременности, а также парентеральное заражение при переливании донорской крови, взятой от паразитоносителей. В эндемических очагах к малярии в большей степени восприимчивы дети и приезжие. Пик заболеваемости малярией совпадает с сезоном активности комаров и приходится на летне-осеннее время.
Пароксизмальный характер лихорадочных приступов при малярии связан с эритроцитарной фазой развития малярийного плазмодия. Развитие лихорадки совпадает с распадом эритроцитов, высвобождением в кровь мерозоитов и продуктов их обмена. Чужеродные для организма субстанции оказывают общетоксическое воздействие, вызывая пирогенную реакцию, а также гиперплазию лимфоидных и ретикулоэндотелиальных элементов печени и селезенки, приводя к увеличению этих органов. Гемолитическая анемия при малярии является следствием распада эритроцитов.
Симптомы малярии
В течении малярии выделяют инкубационный период, период первичных острых проявлений, вторичный латентный период и период рецидивов. Инкубационный период при трехдневной малярии и овале-малярии длится 1-3 недели, при четырехдневной - 2-5 недель, при тропической - около 2-х недель. Типичными клиническими синдромами для всех форм малярии служат лихорадочный, гепатолиенальный и анемический.
Через 1-2 часа фаза озноба сменяется жаром, что совпадает с повышением температуры тела до 40-41 °С. Возникают гиперемия, гипертермия, сухость кожи, инъекция склер, жажда, увеличение печени и селезенки. Может отмечаться возбуждение, бред, судороги, потеря сознания. На высоком уровне температура может удерживаться до 5-8 и более часов, после чего происходит профузное потоотделение, резкое снижение температуры тела до нормального уровня, что знаменует собой окончание приступа лихорадки при малярии. При трехдневной малярии приступы повторяются каждый 3-й день, при четырехдневной – каждый 4-й день и т. д. К 2-3-й неделе развивается гемолитическая анемия, появляется субиктеричность кожи и склер при нормальной окраске мочи и кала.
Своевременное лечение позволяет остановить развитие малярии после 1-2 приступов. Без специфической терапии продолжительность трехдневной малярии составляет около 2 лет, тропической - около 1 года, овале-малярии - 3-4 года. В этом случае после 10-14 пароксизмов инфекция переходит в латентную стадию, которая может длиться от нескольких недель до 1 года и дольше. Обычно через 2-3 месяца видимого благополучия развиваются ранние рецидивы малярии, которые протекают так же, как острые проявления болезни. Поздние рецидивы возникают через 5-9 месяцев - в этот период приступы имеют более легкое течение.
Осложнения малярии
Тяжелыми, порой жизнеугрожающими осложнениями малярии могут служить малярийная кома, малярийный алгид, разрыв селезенки, отек мозга, ОПН, ДВС-синдром, психические нарушения. Малярийной комой чаще всего осложняется течение тропической малярии. Развитие комы связано с нарушениями микроциркуляции головного мозга в результате образования паразитарных тромбов, состоящих из эритроцитов, зараженных шизонтами. В течении малярийной комы выделяют периоды сомноленции (сонливость, адинамия), сопора (резкая заторможенность, снижение рефлексов) и глубокой комы (отсутствие сознания и рефлексов). Летальный исход при возникновении данного осложнения наступает в 96-98% случаев.
Малярийный алгид сопровождается развитием коллаптоидного состояния с артериальной гипотонией, нитевидным пульсом, гипотермией, снижением сухожильных рефлексов, бледностью кожных покровов, холодным потом. Нередко возникают поносы и явления дегидратации. Признаки разрыва селезенки при малярии возникают спонтанно и включают в себя кинжальную боль в животе с иррадиацией в левое плечо и лопатку, резкую бледность, холодный пот, снижение АД, тахикардию, нитевидный пульс. По данным УЗИ выявляется свободная жидкость в брюшной полости. При отсутствии экстренного оперативного вмешательства быстро наступает смерть от острой кровопотери и гиповолемического шока.
Отек мозга развивается при злокачественной, молниеносной форме трехдневной малярии, чаще у детей-дошкольников и подростков. Возникает на высоте лихорадочного пароксизма и характеризуется сильной головной болью, судорогами с потерей сознания, выделением пены изо рта и скорой гибелью пациента. Развитие острой почечной недостаточности при малярии связано с массивным внутрисосудистым гемолизом эритроцитов, нарушением почечного кровообращения, интенсивной гемоглобинурией. Обычно является исходом гемоглобинурийной лихорадки. Специфическим осложнением тропической малярии выступают психические расстройства, включающие в себя психомоторное возбуждение, бред, галлюцинации и т. д.
Диагностика малярии
Фундамент клинической диагностики малярии составляет триада признаков: приступообразная интермиттирующая лихорадка, повторяющаяся каждые 48 или 72 часов, гепатоспленомегалия, гемолитическая анемия. Одновременно выясняется факт посещения больным эндемичных регионов, перенесенных гемотрансфузий и парентеральных вмешательств в течение последних 2-3-х месяцев.
Специфическим лабораторным методом диагностики малярии служит микроскопия толстой капли крови, позволяющая обнаружить наличие и количество паразитов. Качественную идентификацию вида плазмодия и стадию шизогонии проводят путем исследования на малярийный плазмодий мазка крови. Забор крови лучше производить на высоте лихорадочного приступа. Вспомогательную роль в выявлении малярии играют серологические методы – РИФ, РФА, РНГА. В плане дифференциальной диагностике наибольшее значение имеет исключение у лихорадящего больного бруцеллеза, возвратного тифа, лейшманиоза, сепсиса, туберкулеза, менингоэнцефалита, гемолитической желтухи, цирроза печени, лейкоза и др.
Лечение малярии
Больные с подозрением на малярию госпитализируются в инфекционный стационар с назначением строгого постельного режима, обильного питья, инфузионной терапии, общеукрепляющего и симптоматического лечения. При необходимости больным проводится гемосорбция и гемодиализ.
Первоначально для специфической химиотерапии малярии использовался хинин, выделенный из коры хинного дерева. В настоящее время создано большое количество синтетических препаратов, однако из-за быстрого развития резистентности паразитов к синтетическим лекарствам, хинин до сих пор не утратил своей актуальности. В зависимости от оказываемого действия противомалярийные препараты делятся на тканевые шизонтоциды, воздействующие на тканевые формы малярийного плазмодия (хиноцид, примахин) и гематоциды, воздействующие на эритоцитарные формы возбудителя (хлорохин, пириметамин, мепакрин, хинин и др.). Они назначаются в различных сочетаниях и по определенной схеме в зависимости от формы и тяжести течения малярии. Так, при трехдневной малярии обычно проводится 3-дневный курс лечения хлорохином, затем 10-дневный прием примахина или хиноцида для уничтожения тканевых форм паразита. Возможны и другие схемы противомалярийной терапии.
Прогноз и профилактика малярии
Своевременная и правильная терапия малярии приводит к быстрому купированию клинических проявлений. Летальные исходы при проведении лечения возникают примерно в 1% случаев, как правило, при осложненных формах тропической малярии.
Профилактика малярии проводится в двух направлениях: уничтожение комаров-переносчиков возбудителей и индивидуальная защита. Первое направление включает обработку территорий инсектицидами. Второе - использование средств индивидуальной защиты (кремов, лосьонов, противомоскитных сеток), проведение специфической химиопрофилактики лицам, совершающим поездки в районы, неблагополучные по малярии. С целью раннего выявления больных и паразитоносителей всем пациентам с лихорадкой неясного генеза должна проводиться микроскопия крови на малярию.
Читайте также: