
Подострый склерозирующий панэнцефалит корь
Обновлено: 12.05.2024
Подострый склерозирующий панэнцефалит чаще встречается у детей, перенесших корь до двухлетнего возраста. Подострый склерозирующий панэнцефалит развивается через несколько лет после кори и обычно за несколько месяцев приводит к деменции. Для него характерен очень высокий титр противокоревых антител в сыворотке и спинномозговой жидкости. По-видимому, это заболевание является результатом реакции макроорганизма на нарушенную репликацию вируса кори в головном мозге.
В США ежегодно регистрируется менее 10 случаев этой редкой болезни. Заболеваемость резко снизилась после введения противокоревой вакцинации. Анамнез в большинстве случаев типичен: корь в раннем детстве (до 2 лет), латентный период в течение 6-8 лет, затем нарастающие неврологические расстройства; в 85% случаев диагноз ставится в возрасте 5-15 лет.
Что провоцирует / Причины Подострого склерозирующего панэнцефалита:
В этиологии подострых склерозирующих энцефалитов большую роль играет персистирующая вирусная инфекция, по-видимому, коревая. У больных подострым склерозирующим панэнцефалитом обнаруживаются в крови и цереброспинальной жидкости очень высокие титры коревых антител (не отмечающиеся даже у больных острой коревой инфекцией). Выявлен также специфический коревой иммуноглобулин, свойственный текущей коревой инфекции. Вирус кори в головном мозге находится в супрессированном состоянии. Некоторым исследователям удалось выделить вирус кори из ткани мозга умерших больных. В патогенезе заболевания играют роль аутоиммунные механизмы, а также приобретенный или врожденный дефект иммунной системы.
Патогенез (что происходит?) во время Подострого склерозирующего панэнцефалита:
В патогенезе подострого склерозирующего панэнцефалита имеют значение два основных фактора - персистенция вируса в ЦНС и нарушения иммунологической реактивности.
Микроскопически обнаруживаются выраженная диффузная демиелинизация и глиоз белого вещества полушарий большого мозга. В ряде случаев имеется множество глиозных узелков. В других случаях обнаруживаются оксифильные включения в ядрах нейронов коры, подкорки, ствола мозга на фоне их дистрофических изменений. Осевые цилиндры сначала остаются относительно интактными, затем гибнут. Отмечается умеренно выраженная периваскулярная инфильтрация лимфоидными и плазматическими клетками. Для лейкоэнцефалита Шильдера характерно разрастание глии с очагами склероза.
Симптомы Подострого склерозирующего панэнцефалита:
В поздней стадии болезни возникают моно-, геми- и тетрапарезы спастического характера, которые как бы накладываются на екстрапирамидные и лобно-мозжечковые двигательные нарушения. Выявляются сенсорная и моторная афазия, слуховая и зрительная агнозия. Прогрессирует кахексия.
Течение и прогноз
В течении подострых склерозирующих энцефалитов выделяют четыре стадии.
- В 1-й стадии ведущими симптомами являются изменения личности, отклонения в поведении, нарастающие дефекты высших мозговых функций, разнообразные гиперкинезы, судорожные и несудорожные припадки. Эта I стадия (психотическая) длится от 1 до 12 мес.
- Во 2-й стадии нарастают экстрапирамидные нарушения тонуса и расстройства вегетативной центральной регуляции; Нарушения мышечного тонуса нередко носят смешанный спастико-ригидный характер, часто отмечаются гиперкинезы (атетоз, миоклонии, дрожание). Подкорковые нарушения в начальном периоде развиваются по моно- или гемитипу, но в дальнейшем распространяются на обе руки и ноги. Симптомы поражения пирамидных путей при панэнцефалите появляются на фоне уже развернутой картины болезни. К часто встречающимся симптомам относятся также статическая и локомоторная атаксия (вследствие поражения мозжечка или лобной доли коры). К типичным и постоянным признакам болезни относятся эпилептические припадки в виде генерализованных судорог, простых и сложных абсансов, сумеречных состояний и автоматизмов. Эта стадия обычно продолжается от 6 мес до 1 года.
- 3-я стадия характеризуется резким нарастанием мышечной ригидности, появлением торсионных спазмов, миогенных контрактур. У больных отмечаются продолжительные тонические судороги децеребрационного типа, нарастает слабоумие. В связи с усилением ригидности ослабевают клонические судороги и гиперкинезы. Длительность III стадии - несколько месяцев.
- Для последней (коматозной) стадии подострых склерозирующих энцефалитов характерны полная утрата психических функций, стойкая децеребрационная ригидность, гипертермические кризы с лихорадкой гектического типа, кахексия. Летальный исход наступает при развитии циркуляторного коллапса или интеркуррентного заболевания.
Течение склерозирующих энцефалитов неуклонно прогрессирующее и всегда заканчивается летально. Длительность заболевания обычно от 6 мес. до 2-3 лет. Встречаются формы, протекающие хронически с периодически наступающими ремиссиями. Смерть наступает в состоянии полной обездвиженности, кахексии, маразма, чаще всего в эпилептическом статусе или вследствие пневмонии.
Клиническая картина лейкоэнцефалита Шильдера имеет некоторые особенности: больше выражены пирамидные симптомы, доминирующие над экстрапирамидными, чаще отмечаются большие эпилептические припадки. В начальных стадиях превалируют психические нарушения. Возможно течение в виде псевдотуморозной формы с признаками нарастания одноочаговых полушарных симптомов, сопровождающихся внутричерепной гипертензией. Характерно поражение черепных нервов, особенно II и VIII пар. Возможна амблиопия вплоть до амавроза. На глазном дне обнаруживается атрофия дисков зрительных нервов. В некоторых случаях при амаврозе остаются сохранными зрачковые реакции на свет, что обусловлено центральным характером (за счет поражения затылочной доли) амавроза. В цереброспинальной жидкости обнаруживаются умеренный плеоцитоз, повышение содержания белка и уровня гамма-глобулина. Коллоидная реакция Ланге дает паралитическую кривую при подострых склерозирующих энцефалитах, воспалительную и смешанную - при лейкоэнцефалите Шильдера. Патологические изменения реакции Ланге и гипергаммаглобулиноррахия являются ранними признаками лейко- и панэнцефалита. Обнаруживается повышение уровня олигоклонального ликвора. В сыворотке крови и цереброспинальной жидкости определяются чрезвычайно высокие (особенно при подострых склерозирующих энцефалитах) титры коревых антител. На ЭЭГ регистрируются периодические стереотипные регулярные билатерально-синхронные высокоамплитудные разряды электрической активности (комплексы Радемеккера). При эхоэнцефалоскопии, проводимой в случаях с псевдотуморозным течением лейкоэнцефалита, смещения срединных структур не обнаруживается. Наиболее информативна компьютерная аксиальная томография.
Диагностика Подострого склерозирующего панэнцефалита:
При ЭЭГ регистрируются характерные изменения: периодические (каждые 3-8 с) разряды высокоамплитудных остроконечных медленных волн, чередующиеся с периодами подавления активности. Характерная картина спинномозговой жидкости - отсутствие клеток, нормальная или слегка повышенная концентрация белка и очень высокая относительная концентрация гамма-глобулинов (более 20% всего белка спинномозговой жидкости). Неизменно повышен титр противокоревых антител в спинномозговой жидкости, часто имеются специфические олигоклональные иммуноглобулины.
При КТ и МРТ видны множественные очаги поражения белого вещества, атрофия коры и необструктивная гидроцефалия.
Особыми методами можно выделить вирус кори из культуры ткани мозга. Вирусные антигены выявляют иммуноцитохимически, а вирусную РНК - методами флюоресцентной гибридизации in situ или ПЦР.
Лечение Подострого склерозирующего панэнцефалита:
К патогенетическим методам терапии относится применение глюкокортикоидов, однако довольно часто это лечение малоэффективно. Для купирования судорог используют барбитураты (фенобарбитал, бензонал, гексамидин) в сочетании с карбамазепином (финлепсин, тегретол) или производными вальпроевой кислоты (депакин, конвулекс) в возрастных дозах. Для снижения мышечного тонуса применяют мидокалм, баклофен (лиоресал). Назначают сосудорасширяющие препараты (кавинтон, сермион), ноотропил, витамины группы В, в отдельных случаях анаболические гормоны (нераболил, ретаболил), общеукрепляющие средства.
Есть данные о том, что инозин пранобекс (100 мг/кг/сут внутрь) иногда позволяет продлить жизнь и улучшить состояние, но они противоречивы.
К каким докторам следует обращаться если у Вас Подострый склерозирующий панэнцефалит:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Подострого склерозирующего панэнцефалита, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Подострый склерозирующий панэнцефалит — медленная инфекция, вызванная вирусом кори. Латентный период составляет до 8 лет. Подострый коревой панэнцефалит дебютирует расстройствами личности и поведения, затем появляются прогрессирующие мышечно-тонические, двигательные, зрительные, когнитивные нарушения, пароксизмальные состояния. Основные методы диагностики: электроэнцефалография, КТ/МРТ головного мозга, анализ цереброспинальной жидкости, выявление антител к кори в крови, ликворе. Этиотропная терапия осуществляется антивирусными фармпрепаратами, симптоматическое лечение проводится противоэпилептическими средствами.
МКБ-10
Общие сведения
Причины ПСП
Патология является классической медленной инфекцией ЦНС. Инфекционным агентом выступает вирус кори, сохраняющийся в организме в персистирующем состоянии после перенесённой естественной коревой инфекции. В большинстве случаев ПСП наблюдается у детей, переболевших корью в возрасте до двух лет. Латентный период продолжается 6-8 лет, по истечении которых развивается быстро прогрессирующий коревой панэнцефалит. Факторы, обуславливающие вирусную персистенцию, остаются неизвестными. Отдельные авторы предполагают, что в основе лежит изменённая иммунологическая реактивность организма, обуславливающая неполную элиминацию вируса.
Патогенез
Этиопатогенетические механизмы не изучены, вызывающие активацию вируса триггеры не определены. После многолетнего латентного периода персистирующие в церебральных клетках вирусы кори начинают активную репликацию, что обуславливает тотальные воспалительные изменения тканей головного мозга — подострый панэнцефалит. В воспалительный процесс вовлекаются белое вещество, подкорковые ганглии, кора, мозговые оболочки. Очаги поражения распределяются неравномерно, в них происходит гибель нейронов, компенсаторно разрастается глиальная ткань. На поздних стадиях склерозирующий панэнцефалит характеризуется диффузной демиелинизацией в сочетании с глиозом. Микроскопически в поражённых нейронах обнаруживаются специфические включения. Исследование при помощи меченых антител выявляет в них наличие антигенов коревого вируса.
Классификация
Клиническая симптоматика ПСП весьма вариабельна, однако у всех пациентов прослеживается стадийное течение. Понимание фазы заболевания имеет клиническое и прогностическое значение, в связи с чем подострый склерозирующий панэнцефалит классифицируют на 4 основные стадии:
- I стадия (начальная, психотическая) характеризуется нарастающими изменениями в характере, поведении, интеллектуальных способностях больного. До появления мышечно-тонических нарушений часто ошибочно диагностируется как психиатрическая патология. Продолжается 2-12 месяцев.
- II стадия начинается с появления двигательных расстройств (гиперкинезов), пароксизмальных эпизодов (судорожных приступов, абсансов, атонических падений). В дальнейшем присоединяется различная неврологическая симптоматика. Стадия продолжается 6-12 месяцев.
- III стадия протекает с быстрым прогрессированием деменции, нарастанием мышечной ригидности, ослаблением судорожного синдрома. Длится несколько месяцев.
- IV стадия (терминальная, коматозная) — полный распад психических функций, децеребрационная ригидность, кахексия. Пациенты впадают в кому с последующим летальным исходом.
Симптомы ПСП
Подострый склерозирующий панэнцефалит дебютирует постепенно усугубляющимися отклонениями в поведении: больной становится неряшлив, упрям, агрессивен, раздражителен, равнодушен к окружающим. Начинают преобладать примитивные качества: жадность, эгоизм. Появляются психопатоподобные реакции, нарушения сна, у школьников возникают сложности в обучении. К концу начального периода наблюдаются прогрессирующие мнестические расстройства, интеллектуальное снижение, нарушения речи (дизартрия, афазия).
Постепенно гиперкинезы сменяются симптоматикой паркинсонизма, смешанные изменения тонуса переходят в тотальную ригидность. Экстрапирамидные расстройства сочетаются с вегетативной симптоматикой: повышенной сальностью кожи, гипергидрозом, лабильностью давления, гиперсаливацией. Нарастающая децеребрационная ригидность приводит к постепенному исчезновению судорожных пароксизмов. Отмечается полный распад личности, насильственный смех/плач, гипертермические кризы, расстройства глотания, дыхания. В терминальной стадии конечности пациента согнуты, имеются сгибательные контрактуры, продуктивный контакт отсутствует, угнетение сознания доходит до стадии комы, возникают трофические изменения тканей.
Осложнения
Прогрессирование зрительных нарушений приводит к амаврозу. Лежачее положение больного на последних стадиях ПСП может вызвать образование пролежней. Инфицирование последних ведёт к местным воспалительным изменениям, попадание инфекции в кровоток — к возникновению сепсиса. Обездвиженность пациента, дыхательные расстройства центрального генеза способствуют возникновению застойной пневмонии. Дисфагия опасна попаданием пищи в дыхательные пути с развитием аспирационной пневмонии. Перечисленные инфекционные осложнения выступают наиболее частыми причинами гибели больных.
Диагностика
Полиморфизм, неспецифичность симптоматики, изолированные психотические проявления начального периода, редкая встречаемость обуславливают позднее диагностирование заболевания. В ходе диагностики невролог опирается на анамнестические сведения о перенесённой в детстве кори, изменения неврологического статуса, данные ЭЭГ и нейровизуализации, результаты исследований на противокоревые антитела. Биопсия головного мозга не показательна, поскольку мозаицизм церебрального поражения делает возможным забор материала из неизменённого участка мозговых тканей. Перечень необходимых дополнительных обследований включает:
- Электроэнцефалографию. Типична картина повышенной медленноволновой активности, возникающая с интервалом 6-8 с и чередующаяся с периодами сниженной биоэлектрической активности. Регистрируемые комплексы носят двусторонний характер, симметричны, синхронны.
- Нейровизуализацию. Проводится методами компьютерной или магнитно-резонансной томографии. МРТ, КТ головного мозга диагностируют диффузное множественное поражение церебральных тканей, атрофические изменения коры, в ряде случаев — гидроцефалию. Изменения в белом веществе наблюдаются спустя 4 месяца от дебюта ПСП. В 50% случаев поражения базальных ганглиев определяются уже на 2-ой стадии.
- Исследование цереброспинальной жидкости. Ликвор для анализа получают путём люмбальной пункции. Исследование выявляет умеренное повышение концентрации белка со значительным нарастанием относительного содержания гамма-глобулинов, наличие специфических олигоклональных IgG. Резкое нарастание титра антикоревых антител свидетельствует о коревой этиологии поражения.
- Анализ крови на противокоревые антитела. Определяет повышение антител в сыворотке до 1:4 — 1:128. В норме титр составляет 1:200 – 1:500. Выявление повышенного титра в крови и цереброспинальной жидкости является важнейшим диагностическим признаком.
Выделение вируса кори из поражённых тканей мозга редко применяется в клинической практике по причине сложности выполнения. В начальной стадии подострый склерозирующий панэнцефалит необходимо дифференцировать от психических расстройств: неврастении, шизофрении, истерии. В последующем дифдиагноз осуществляется с опухолью головного мозга, прогрессирующим краснушным панэнцефалитом, клещевым энцефалитом, болезнью Крейтцфельдта-Якоба.
Лечение ПСП
Специфическая терапия не разработана. Большое значение имеет правильный уход за больным, предупреждение инфекционных осложнений. Проводится этиотропное лечение противовирусными средствами (рибавирин, инозин пранобекс), препаратами интерферона, но оно малорезультативно. В качестве симптоматической терапии назначают антиконвульсанты, эффективные в отношении миоклоний (диазепам, производные вальпроевой кислоты). Для снятия спастического гипертонуса применяют миорелаксанты (толперизон, баклофен). Нарушения дыхания на заключительных стадиях заболевания являются показанием к переводу пациентов на ИВЛ. Тщательный уход и симптоматическое лечение позволяют продлить жизнь больного.
Прогноз и профилактика
В 80% случаев подострый склерозирующий панэнцефалит имеет длительность 1-3 года и приводит к летальному исходу. У 10% пациентов наблюдается молниеносное течение. В 10% случаев удаётся увеличить продолжительность жизни больного, иногда до 10 лет. Наилучшими превентивными мерами, позволяющими предупредить возникновение ПСП, являются мероприятия по предупреждению заболевания корью. Специфическая профилактика проводится путём плановой вакцинации против кори детей в возрасте 12 месяцев.
Прогрессирующий краснушный панэнцефалит — редкая медленная вирусная инфекция ЦНС, обусловленная вирусом краснухи. Манифестирует в период от 8 до 19 лет. Проявляется прогрессирующим умственным снижением, мозжечковым синдромом, спастическим тетрапарезом, нарушениями зрения, эпилептическими пароксизмами. Диагностируется на основании анамнестических и клинических данных, картины церебральной МРТ/КТ, результатов анализа спинномозговой жидкости. Лечение осуществляется симптоматическими средствами, имеет малую эффективность. Заболевание склонно к неуклонному прогрессированию.
Общие сведения
Причины ПКП
Этиофактором заболевания выступает краснушный вирус. Медленная инфекция развивается при заражении в результате:
- Внутриутробного инфицирования. Краснуха при беременности у женщины может протекать бессимптомно. Внутриутробная инфекция плода в первом триместре приводит к множественным порокам развития, далее вероятность аномалий уменьшается. Признаки врождённой краснухи (раннего краснушного энцефалита) обычно наблюдаются в ближайшем периоде после родов. Прогрессирующий краснушный панэнцефалит развивается, если вирус не проявляется сразу, а остаётся в латентном состоянии.
- Краснухи, перенесённой в детстве. В исключительных случаях после перенесённой инфекции не происходит полная элиминация вируса, он персистирует в организме в латентной форме.
К группе риска инфицирования корью в период беременности относятся женщины с низкой концентрацией специфических противокоревых иммуноглобулинов (10-25 МЕ/мл), осуществляющих защиту организма от краснушного вируса.
Патогенез
Симптомы ПКП
Прогрессирующий краснушный панэнцефалит дебютирует неспецифической симптоматикой. Близкие начинают замечать перепады настроения, изменения в характере, поведении больного. Ребёнок хуже учится в школе, возникают проблемы с сосредоточением внимания, запоминанием, усваиванием нового материала. Со временем умеренные когнитивные нарушения перерастают в прогрессирующий интеллектуальный дефицит с исходом в деменцию. Отличительной особенностью ПКП является тяжёлая мозжечковая атаксия, включающая шаткость в положении стоя, неустойчивость во время ходьбы, несоразмерные крупноразмашистые движения, промахивание при выполнении целенаправленных действий.
Развёрнутая стадия заболевания характеризуется эпилептическими пароксизмами, присоединением очагового неврологического дефицита. Наблюдается выраженная атаксия (дискоординация, шаткость), зрительные расстройства, парез лицевого нерва. Типичны пирамидные симптомы: слабость мышц, спастическое повышение мышечного тонуса, повышение сухожильных рефлексов, появление патологических стопных знаков. Двигательные нарушения распространяются на все конечности, развивается тетрапарез. В некоторых случаях наблюдаются миоклонии — непроизвольные сокращения отдельных мышечных групп. Возникает сенсомоторная афазия — речь становится замедленной, упрощённой, непонятной из-за перестановки слогов, нарушается понимание услышанной речи.
В терминальной стадии спастический тетрапарез приковывает пациентов к постели, отсутствует произвольный контроль тазовых органов. Отмечается выраженная деменция, полное отсутствие речи (мутизм), расстройство движений глазных яблок (офтальмоплегия). Нередко наступает кома. Заболевание оканчивается летальным исходом вследствие тотального поражения головного мозга.
Диагностика
- Неврологический осмотр. В ходе обследования невролог устанавливает наличие фациального пареза, пирамидной симптоматики, мозжечкового синдрома, выраженного когнитивного снижения, сенсомоторной афазии.
- Люмбальную пункцию. Процедура позволяет получить образец цереброспинальной жидкости, провести его анализ. Характерно умеренное повышение лимфоцитов (40 кл/мкл), белка (1,5-1,5 г/л) с преобладанием гаммаглобулина. Резко увеличена концентрация специфических антикраснушных антител, что свидетельствует об их продукции в тканях ЦНС.
- Электроэнцефалографию. Не обнаруживает специфических отклонений. Типично диффузное замедление основного ритма.
- Томографию. МРТ, МСКТ и КТ головного мозга выявляют гидроцефалию, генерализованную атрофию церебральных тканей, расширение борозд мозговой коры. Особенно грубо атрофирован мозжечок.
- Анализ крови на противокраснушные антитела. Осуществляется иммуноферментным методом (ИФА). Титр антител повышен, но не столь значительно, как в цереброспинальной жидкости.
Выделить краснушный вирус из полученных путём биопсии церебральных тканей, как правило, не удаётся. Необходимо дифференцировать прогрессирующий панэнцефалит краснушной этиологии от других медленных инфекций, вирусного энцефалита, рассеянного энцефаломиелита. Дифференциальный диагноз проводят с подострым склерозирующим панэнцефалитом, отличающимся нормальной картиной цереброспинальной жидкости с повышением титра противокоревых антител.
Лечение ПКП
Специфическая терапия не разработана. Попытки лечения противовирусными препаратами (амантадином), метаболическими средствами (инозином) оказались безуспешными. Проводится симптоматическая терапия (антиконвульсанты, антигипоксанты, противоотечные средства), однако она не способна приостановить прогрессирование симптомов.
Прогноз и профилактика
Прогрессирующий краснушный панэнцефалит оканчивается летально. Продолжительность заболевания с момента манифестации симптомов не превышает 3-х лет. В связи с отсутствием эффективной терапии большое значение придаётся профилактике инфицирования краснухой беременных женщин и детей. Специфическая профилактика осуществляется путём вакцинации против краснухи детей в возрасте 1 года с последующей ревакцинацией в 6 лет. Не привитым ранее и не переболевшим краснухой 13-летним девочкам рекомендовано однократное введение вакцины. Планирующим беременность женщинам с низким титром противокраснушных антител показана вакцинация по индивидуальным показаниям. Ведение беременности у женщин из группы риска должно осуществляться с контролем титра специфических антител в крови.

Подострый склерозирующий панэнцефалит (ПСПЭ) — прогрессирующее нейродегенеративное, обычно смертельное заболевание ЦНС. Оно вызывается вирусом кори и относится к медленным вирусным инфекциям. Заболевание начинается спустя годы (обычно до 8 лет) после перенесенной кори.
Заболеваемость в развитых странах составляет 1 случай на 1 млн населения, в Индии — до 21 на 1 млн, что связано с уровнем доступности медицинской помощи. Имеющиеся эпидемиологические данные говорят о том, что вакцинация против кори обладает прямым защитным эффектом против ПСПЭ. Чаще поражаются мальчики и мужчины, обычно в возрасте до 20 лет.
Патогенез
В данный момент существует две теории относительно этиологического агента ПСПЭ: согласно одной, заболевание вызывает мутантный вирус кори, также названный вирусом подострого склерозирующего панэнцефалита; возможно, однако, что все патогенные вирусы кори способны инфицировать ЦНС, а развитие ПСПЭ в дальнейшем зависит от иммунного ответа хозяина.
Скорее всего, вирус кори попадает в структуры ЦНС во время первого инфицирования корью и персистирует до развития панэнцефалита. В большей мере поражаются нейроны и олигодендроциты, однако как именно вирус проникает в них — не вполне понятно, ведь эти клетки не экспрессируют известных клеточных рецепторов к вирусу кори. Как показали экспериментальные модели, после проникновения в нейроны вирус может распространяться на соседние структуры транснейронально.
На ранних стадиях в процесс вовлекаются затылочные области, в дальнейшем изменения распространяются на передние отделы коры, и, в последнюю очередь, на подкорковые, стволовые структуры и спинной мозг. На поздних стадиях можно наблюдать картину распространенной деструкции как белого, так и серого вещества головного мозга и признаки атрофии коры.
Гистологически наблюдаются воспалительные изменения в паренхиме и оболочках мозга, демиелинизация, множественные вирусные включения в нейронах, олигодендроцитах и астроцитах, потеря нейронов и астроглиоз.

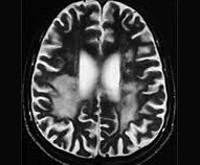
Рисунок 1 | МР-томографическая и МР-спектроскопическая картины мальчика в возрасте 3 лет и 6 месяцев с ПСПЭ.
При поступлении отмечались частые миоклонические судороги. Он перенес корь в возрасте 10 месяцев.
(A) T2-взвешенная аксиальная томограмма: симметричные области гиперинтенсивнности в белом веществе, которые распространяются на подкорковую область. Явления атрофии с выраженными бороздами и расширением желудочков.
(B) МР-спектроскопия: Снижение пика N-ацетиласпартата (NAA), несколько повышен пик холина (Cho).
Клиника
В клиническом течении болезни выделяют 4 стадии:
I стадия характеризуется нарушениями поведения в виде повышенной раздражительности, тревожности, психопатоподобных реакций, падением успеваемости, утомляемостью, расстройствами речи и нарушениями сна. Часто ошибочно диагностируется как психиатрическая патология. Продолжается 2–12 месяцев.
II стадия начинается с появления двигательных расстройств (гиперкинезов), пароксизмальных эпизодов (миоклонических приступов, обычно генерализованных, абсансов, атонических падений). Стадия продолжается 6–12 месяцев.
III стадия протекает с исчезновением миоклонуса, пирамидными и экстрапирамидными расстройствами, прогрессированием деменции, постепенным ослаблением судорожного синдрома. Длится несколько месяцев.
IV стадия (терминальная) — децеребрационная ригидность, декортикация, вегетативное состояние.
В половине случаев возникают офтальмологические изменения в виде некротизирующего ретинита. Также нередки нарушения зрения в виде корковой слепоты.
Диагностические критерии Dyken для диагностики подострого склерозирующего панэнцефалита
- Типичное клиническое течение с прогрессирующими изменениями психического статуса и стереотипными генерализованными миоклоническими приступами
- Характерные изменения ЭЭГ (в виде стереотипных периодических высокоамплитудных дизритмических комплексов, которые связаны с миоклоническими судорогами.
- Повышение отношения содержания глобулинов к уровню альбумина более 20 % в ЦСЖ.
- Возрастание титров антител к вирусу кори в ЦСЖ.
- Типичные гистопатологические находки (а также результаты ПЦР) при исследовании биоптата/аутопсии.
Лучевые признаки
На ранних стадиях процесса может не выявляться никаких изменений. МРТ является методом выбора.
Сигнальные характеристики
- пораженные области демонстрируют повышение МР сигнала на T2ВИ/FLAIR изображениях;
- нередко наблюдается также их контрастное усиление в T1ВИ после внутривенного контрастирования;
- атрофия головного мозга проявляет себя выраженностью борозд, истончением извилин и равномерным компенсаторным расширением желудочков;
- при проведении МР-спектроскопии может обнаруживаться:
— отсутствие или снижение пика N-ацетиласпартата;
— повышение пика холина и мио-инозитола;
— повышение пика лактата;

(A) На аксиальном Т2ВИ отмечается ассиметричные области гиперинтенсивности в обеих гемисферах, а также в правом таламусе и бледном шаре.
(B) Корональное FLAIR изображение демонстрирует вовлечение в процесс моста с распространением на средние ножки мозжечка.

(A) DWI (b = 1000 с/мм²) показывает слегка гиперинтенсивный сигнал от пораженных областей.
(B) ADC карта: повышение интенсивности и повышение значений ADC от пораженных областей (при измерении: 1,17, 1,18, 1,24, 1,49, 1,27, и 1,21 × 10 −3 мм²/с).
Излюбленная локализация изменений: перивентрикулярное и субкортикальное белое вещество; базальные ядра, мозолистое тело, таламусы и спинной мозг поражаются реже. С прогрессированием процесса появляются признаки атрофии полушарий, мозжечка и продолговатого мозга, вплоть до тотальной потери белого вещества на терминальной стадии. Серое вещество вовлекается в сравнительно меньшей мере.
Помните, что выраженность лучевых проявлений может не коррелировать с клинической картиной.

МР-спектроскопия показала снижение пика N-ацетиласпартата (при сравнении с креатином) при нормальном пике холина.
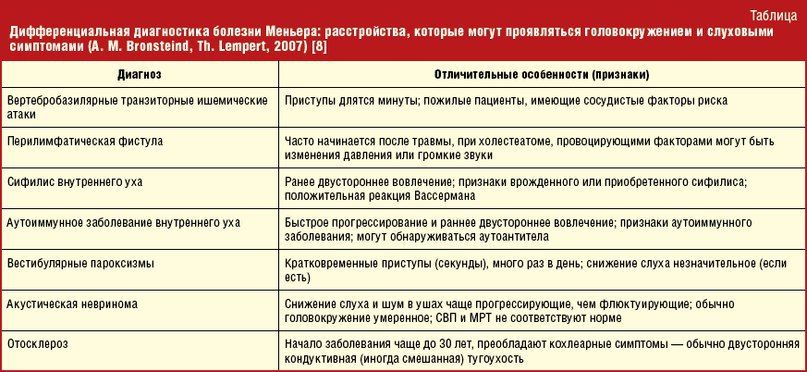
Дифференциальная диагностика
Диагностика этой крайне редкой патологии в начальной стадии представляет большие трудности. В ретроспективном исследовании, включавшем данные о 307 пациентах, отмечено, что в 78,8 % случаев при первичном обращении выставлялся другой диагноз: эпилепсия, абсансная эпилепсия, синдром Леннокса-Гасто, метахроматозная лейкодистрофия, болезнь Вильсона-Коновалова, васкулит, спиноцеребеллярная атаксия, кататоническая шизофрения, гемипаркинсонизм, болезнь Шильдера и даже симуляция. Лист дифференциальных диагнозов с разными проявлениями ПСПЭ можно увидеть в таблице 1.

Таблица 1 | Лист дифференциальных диагнозов при диагностике ПСПЭ.
Лечение
Специфическое лечение на данный момент не разработано.
Изопринозин был одним из первых препаратов, показавших эффективность в стабилизации болезни. Однако во многих других исследованиях положительное действие препарата оказалось не доказанным. Разные исследователи применяли также интравентрикулярное введение альфа-интерферона, бета-интерферона и рибавирина, а также комбинации двух и трех препаратов. Пока, однако, не накоплено достаточно данных об их эффективности.
В качестве симптоматической терапии назначают антиконвульсанты, эффективные в отношении миоклоний (диазепам, производные вальпроевой кислоты). Для снятия спастического гипертонуса применяют миорелаксанты (толперизон, баклофен). Нарушения дыхания на заключительных стадиях заболевания являются показанием к переводу пациентов на ИВЛ.

Головокружение, являясь крайне распространенной жалобой при первичном осмотре, нередко не рассматривается врачами как важный симптом. Однако за ним может скрываться очень грозная патология, такая как болезнь Меньера (idiopathic endolymphatic hydrops, или идиопатическая эндолимфатическая водянка, лабиринтная водянка).
Что же включает в себя данная патология?
По определению, болезнь Меньера является идиопатической. Другими словами, если причина известна, патологический процесс уже не может быть назван болезнью Меньера. Тем не менее, если корень проблемы заключается в повышении эндолимфатического давления, стоит рассмотреть и другие причины гидропса. Болезнь Меньера в таких случаях следует отличать от этих причин.
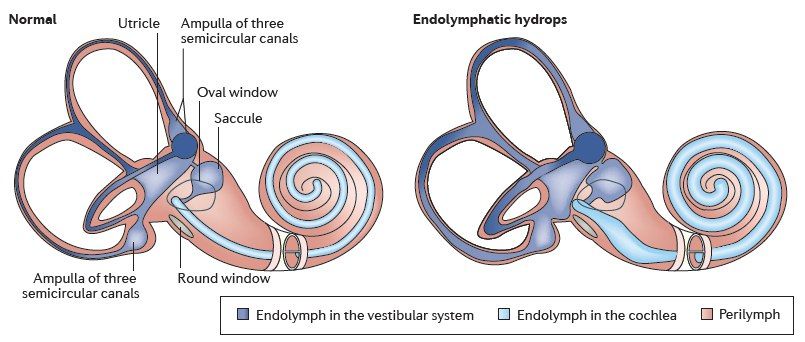
Существует несколько теорий, пытающихся объяснить причину возникновения данной патологии:
Аутоиммунные заболевания, такие как волчанка и ревматоидный артрит, могут вызывать воспалительную реакцию в пределах лабиринта. Аутоиммунная этиология была подтверждена после того, как было установлено, что у пациентов с БМ определяются аутоантитела к щитовидной железе, а также иммунные комплексы в эндолимфатическом мешке (иммунологическая теория).
Заболевания, которые могут привести к повышению эндолимфатического давления, включают метаболические нарушения, гормональный дисбаланс, травмы, а также различные инфекции (например, сифилитический отит и синдром Когана — интерстициальный кератит). Отдельно следует остановиться на метаболических нарушениях. Имеется предположение, что повышение уровня калия в эндолимфатическом пространстве вызывает повреждение волосковых клеток и вестибулярного эпителия, что приводит к развитию симптомов головокружения и тугоухости (метаболическая теория).
Кроме того, замечено, что при болезни Меньера частота аллергических реакций выше, чем среди населения в целом (у 50 % установлено наличие аллергии к веществам, передающимся по воздуху). Пищевые триггеры также являются важными факторами при возникновении БМ (у 45 % больных с аллергией на пищевые продукты) — аллергическая теория.
Также выделяют сосудистую теорию (в связи с частым сочетанием БМ и мигрени у больных), генетическую (аутосомно-доминантный тип наследования, участок DFNA9 гена СОСН) и анатомическую (патология височной кости, в результате чего малый по размеру эндолимфатический мешок расположен позади лабиринта).
В двух из трех случаев обращения с жалобами на головокружение или потерю слуха процесс чаще всего является односторонним.
Первыми проявлениями при БМ, как описано выше, могут стать как вестибулярные нарушения (вертиго), так и слуховые (снижение остроты слуха, наличие шума в ушах).
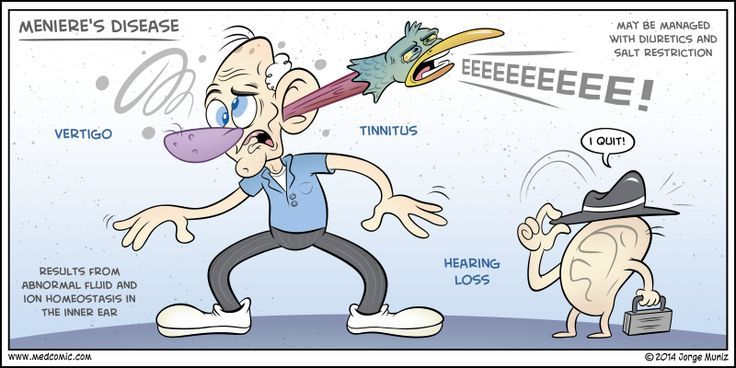
Выделяют 3 формы БМ:
- Классическая (слуховые + вестибулярные нарушения);
- Кохлеарная (слуховые нарушения с постепенным присоединением вестибулярных);
- Вестибулярная (картина манифестации нарушений обратна кохлеарной форме);
При классическом течении БМ выделяют 3 стадии заболевания:
Достоверная (очевидная) БМ по рекомендациям AAO-HNS (Американская академия отоларингологии и хирургии головы и шеи), JSER (Японское общество по изучению равновесия) и EAONO (Европейская академия отологии и нейро-отологии) представляет собой:
- два или более самопроизвольно возникающих приступа головокружения продолжительностью 20 мин и более;
- потеря слуха, подтвержденная данными аудиологических исследований по меньшей мере в одном случае;
- шум в ушах или ощущение заложенности в пораженном ухе.
При исключении других причин, в результате которых может развиться вышеописанная симптоматика.
- проба Ромберга;
- тест Фукуды/маршевая проба;
- указательная проба Барани;
- проба Бабинского-Вейля;
- проба Ринне/Вебера.
В дополнение к этому проводят калорические пробы, вращательный тест Барани и др.
Инструментальная диагностика включает в себя:
- экстра - и транстимпанальную электрокохлеографию (ЭКоГ);
- аудиометрию (тональная пороговая, надпороговая, речевая).
При ЭКоГ измеряется соотношение суммарного потенциала (СП) и потенциала действия (ПД). При БМ — СП/ПД> 0,3.
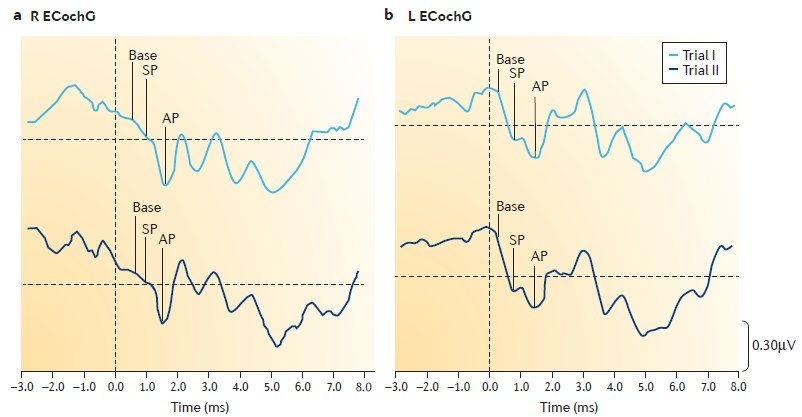
Для верификации диагноза рекомендуется применение дегидратационного теста, при котором после проведения тоновой пороговой аудиометрии больному вводят осмотический диуретик из расчёта 1–1,5 мг на кг массы тела пополам с фруктовым соком и повторяют аудиометрию через 1, 2, 3, 24 и 48 часов.
Основной целью лечения болезни Меньера является оказание помощи при острых приступах головокружения, предотвращение повторных атак и устранение прогрессирующего ухудшения слуха и вестибулярной функции в пораженном ухе (или ушах). Несмотря на прогресс в достижении первых двух целей, устранение прогрессирующего во времени ущерба слуху и вестибулярной функции оказалось недостижимым на данный момент времени. Здесь необходимо напомнить, что БМ часто ассоциируется с такими патологиями, как мигрень, СОАС, аутоиммунными заболеваниями, коагулопатиями/васкулопатиями, поэтому было высказано предположение, что цереброваскулярная ишемия способствует увеличению частоты возникновения атак. Необходимо провести тщательный поиск факторов риска сосудистых заболеваний и по возможности их коррекцию. У людей младше 50 лет мигрень является наиболее распространенным кофактором БМ. С коррекцией приступов мигрени, а также их профилактикой в межприступный период вы можете ознакомиться в нашем обзоре. У людей старше 50 лет, для которых характерна полиморбидность, сопутствующие БМ нозологии также требуют своевременной коррекции ввиду возможного влияния на прогрессирование БМ.
Диуретики, использующиеся в качестве терапии первой линии ввиду их возможности уменьшения объема эндолимфы, в результате которого предотвращается формирование гидропса, также весьма неоднозначны в их назначении в ситуации с БМ. Существуют 2 противоположные группы исследований, одна из которых говорит, что их назначение оправдано, особенно у женщин в период менопаузы. Другие же заявляют, что эффект, полученный от их применения, сравним с плацебо. Но не стоит забывать, что комплекс проводимой терапии должен подбираться индивидуально. В случае БМ наиболее часто назначают комбинацию гидрохлортиазида с триамтереном или диакарб. Касаемо ацетазоламида, его назначение оправдано в ситуациях, когда БМ сочетается с мигренью с аурой, или в качестве терапии второй линии, когда препараты, используемые в качестве профилактики мигрени в межприступный период, не оказывают должного эффекта.
Также необходимо помнить о таком препарате, как бетагистина дигидрохлорид — синтетический аналог гистамина (Н3 подгруппа), который по некоторым данным должен составлять основу терапии острых приступов головокружений. Однако здесь стоит сказать, что существует достаточно большая подборка исследований, свидетельствующая о неоднозначности его применения. Так, в январе 2016 года British Medical Journal (BMJ) на основе данных нескольких Кокрановских обзоров пришел к выводу, что бетагистин вряд ли можно считать эффективным в отношении БМ. Они связали его широкое применение, возможно, с высокой толерантностью в отношении дозирования препарата, низкими рисками развития нежелательных реакций, а также банальным отсутствием альтернативы. В поддержку вышесказанному, в том же 2016 году было проведено многоцентровое исследование BEMED (1450 обследованных пациентов), по результатам которого показано, что назначение как низких (48 мг/сут), так и высоких доз (144 мг/сут) этого препарата не показало значительного влияния на снижение частоты острых приступов головокружений, а также на восстановление слуха и вестибулярных функций по сравнению с контрольной группой. Это еще раз подтверждает неоднозначность назначения данного препарата.
Поэтому в качестве альтернативы бетагистину можно предложить Н1-блокаторы, которые способны проникать через ГЭБ. К ним относят дименгидринат, дифенгидрамин, меклозин.
В ряде случаев оправдано использование седативных средств, например, лоразепама/диазепама при остром приступе головокружения. Симптоматический эффект седативных средств при остром головокружении связан с их общим действием, в условиях которого снижается способность вестибулярных ядер анализировать и интерпретировать импульсы, поступающие из лабиринта.
Хирургическое лечение, согласно критериям AAO-HNS, рекомендуется при неэффективности консервативной терапии в течение 6 месяцев.
Читайте также: