
Прогрессирующая мультифокальная лейкоэнцефалопатия что это такое
Обновлено: 25.04.2024
Научный центр неврологии РАМН, Москва
Прогрессирующая мультифокальная лейкоэнцефалопатия (обзор литературы)
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2012;112(9‑2): 29‑33
Захарова М.Н. Прогрессирующая мультифокальная лейкоэнцефалопатия (обзор литературы). Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2012;112(9‑2):29‑33.
Zakharova MN. Progressive multifocal leukoencephalopathy (review). Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2012;112(9‑2):29‑33. (In Russ.).
Научный центр неврологии РАМН, Москва
Статья посвящена проблеме прогрессирующей мультифокальной лейкоэнцефалопатии (ПМЛ), которая является в большинстве случаев фатальным прогрессирующим демиелинизирующим заболеванием ЦНС. ПМЛ представляет собой оппортунистическую инфекцию, которая развивается в условиях иммуносупрессии, вызываемой как хроническими заболеваниями, так и агрессивной терапией. Рассматриваются возможные механизмы развития ПМЛ, диагностика заболевания и современные подходы к лечению.
Научный центр неврологии РАМН, Москва
Прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ) - это редкое прогрессирующее демиелинизирующее заболевание центральной нервной системы (ЦНС), вызванное реактивацией существующего в латентном состоянии папова вируса (JC-вируса) при иммунодефицитных состояниях [8].
Первое описание двух случаев, похожих на ПМЛ, было сделано в 1930 г. J. Halervorden. Термин ПМЛ был введен в 1958 г. K. Astrom и соавт. [2], которые выделили ПМЛ в самостоятельное заболевание. Первые случаи ПМЛ были описаны ими при лимфопролиферативных заболеваниях - хроническом лимфолейкозе, ходжкинской лимфоме. Предположение о вирусной этиологии заболевания впервые было высказано в работах [23] после идентификации внутриядерных включений в олигодендроглиоцитах.
Вирусная этиология ПМЛ была установлена в 1971 г., когда из мозга больного J. Cunningham был выделен вирус, получивший название по имени этого исследователя - JC-вирус [21]. В 1984 г. было установлено, что JC-вирус является ДНК-содержащим вирусом семейства папова вирусов [10].
В развитии эпидемиологических исследований при ПМЛ можно выделить 4 этапа. До 80-х годов ПМЛ являлась редким заболеванием; заболеваемость составляла 1:1 000 000 человек. С 1958 по 1984 г. всего описано 230 случаев ПМЛ. До эпидемии СПИДа 80% случаев ПМЛ было связано с лимфопролиферативными заболеваниями, ходжкинской лимфомой, тяжелыми формами туберкулеза. С начала 90-х годов (1990-1996), в связи с ростом ВИЧ-инфекции, заболеваемость ПМЛ выросла в 5 раз и в настоящее время составляет 1:200 000 человек в популяции [22]. При этом среди ВИЧ-инфицированных встречаемость ПМЛ до применения высокоактивной антиретровирусной терапии (ВААРТ) составляет 3,3:1000. С внедрением ВААРТ заболеваемость ПМЛ снизилась в 2,5 раза и составила 1,3 на 1000 ВИЧ-инфицированных [9]. С 2005 г. все чаще стали регистрировать случаи ПМЛ у не ВИЧ-инфицированных больных с аутоиммунными заболеваниями, после трансплантации органов, с рассеянным склерозом в результате применения новых методов агрессивной иммуносупрессии (глюкокортикостероиды, пуриновые аналоги - флударибин, кладрибин, азатиоприн, алкилирующие соединения - циклофосфамид, кармустин, декарбазин, моноклональные антитела). Заболеваемость ПМЛ у этих пациентов колеблется от 1:1000 до 1:10 000 [11, 15, 16].
Согласно современным эпидемиологическим данным, к основным состояниям, вызывающим ПМЛ, относятся: ВИЧ/СПИД в 80% случаев, лимфомиелопролиферативные заболевания и злокачественные опухоли в 13% случаев, трансплантация органов и тканей в 5% случаев и аутоиммунные воспалительные заболевания, в том числе системная красная волчанка, склеродермия, ревматоидный артрит, дерматомиозит, которые составляют 2% случаев.
До настоящего времени механизм заражения JC-вирусом неизвестен. Предполагают как воздушно-капельный, так и фекально-оральный пути заражения. Асимптомное инфицирование происходит в ранний период жизни, при этом персистенция вируса наблюдается в CD34 стволовых клетках костного мозга, лимфоидных органах и эпителиальных клетках почек, куда JC-вирус попадает из лимфоцитов периферической крови и миндалин.
В то же время у детей старше 11 лет в крови выявляются специфические антитела к JC-вирусу в 50% случаев, у лиц старше 30 лет - в 80%. У здоровых лиц JC-вирус не вызывает развития ПМЛ, хотя периодически он выделяется методом полимеразной цепной реакции (ПЦР) у 30% - в моче и у 39% - в ткани миндалин [4].
При иммунодефицитном состоянии происходит реактивация JC-вируса с попаданием его в кровь и далее в ЦНС.
В настоящее время доказано, что существуют различные изоформы JC-вируса в гемопоэтической и мочевыделительной системах. При ПМЛ в мозге идентифицирована изоформа JC-вируса, гомологичная вирусу, выделенному из костного мозга, но не из мочи и почечного эпителия.
Генетические исследования позволили идентифицировать ряд изменений регуляторного участка вирусного генома и точковые мутации VPI белка JC-вируса, выделенного из мозга больных ПМЛ в отличие от здоровых лиц [27, 29].
Генетические модификации усиливают аффинность и специфичность JC-вируса к клеточным рецепторам, повышая его вирулентность и трансмиссивность.
Ключевым звеном в предотвращении реактивации вируса и развития ПМЛ является состояние Т-клеточного иммунитета, содержание CD4+ Т-клеток и цитотоксичных CD8+ Т-клеток [18].
Это подтверждается развитием ПМЛ только при иммунодефицитных состояниях, а также прямой связью между содержанием CD4+ и CD8+ Т-клеток и прогнозом у ВИЧ-инфицированных больных ПМЛ. Известно, что глубокая иммуносупрессия (не менее 6 мес) предшествует реактивации JC-вируса.
Основными факторами риска развития ПМЛ являются длительная иммуносупрессия и угнетение Т-клеточного звена иммунитета. Учитывая, что большинство больных ПМЛ (85%) являются
ВИЧ-инфицированными, у этих больных основным предрасполагающим фактором является значительное снижение числа CD4+ Т-клеток (менее 200 клеток/мкл).
При других состояниях количество CD4+ Т-клеток может быть различно, при этом уровень их определяет темп развития ПМЛ. Так, при ПМЛ, вызванной применением ритуксимаба, установлено, что уровень CD4+ Т-клеток определяет интервал между последней дозой препарата и манифестацией клинических проявлений ПМЛ. При содержании CD4+ Т-клеток менее 500 клеток/мкл и выраженном снижении иммуноглобулинов IgG ПМЛ у больных, получающих ритуксимаб, развивается менее чем через 3 мес после последнего введения препарата. При уровне CD4+ Т-клеток более 500 клеток/мкл этот период несколько продолжительнее (в среднем 17 мес).
Уровень CD4+ Т-клеток и снижение индекса CD4+ Т-клеток/CD8+ Т-клеток влияет не только на темп развития ПМЛ, но и на выживаемость. Смертность от ПМЛ у больных с низким уровнем CD4 +Т-клеток (менее 500/мкл) на фоне лечения ритуксимабом составляет 100%, тогда как у остальных больных - 84%.
In vitro JC-вирус способен инфицировать олигодендроциты, астроциты, моноциты, В-лимфоциты, Т-лимфоциты, клетки предшественники гемопоэтических клеток в костном мозге. В настоящее время превалирует мнение о реактивации вируса на периферии и проникновении его через ГЭБ в ткани мозга.
В ЦНС основной мишенью для JC-вируса являются олигодендроциты, с которыми он связывается через серотониновый 5-гидрокситриптамин-2А рецептор на поверхности глиальных клеток. Эти же рецепторы экспрессируются астроцитами, В-клетками, почечным эпителием. JC-вирус вызывает лизис миелинобразующей клетки и, как следствие, массивную демиелинизацию мозговой ткани.
Основными патоморфологическими признаками ПМЛ являются множественные очаги демиелинизации, вызванные гибелью олигодендроцитов, наибольшее число их встречается в полушариях большого мозга, мозговом стволе и мозжечке. Воспалительные изменения в головном мозге практически отсутствуют.
Гистологически при ПМЛ выявляются следующие изменения: измененные олигодендроциты с увеличенными ядрами и внутриядерными вирусными включениями; пролиферация астроцитов с образованием гигантских причудливой формы клеток с гиперхроматическими ядрами; множественные очаги демиелинизации с образованием полостей в них; иногда наблюдаются также изменения в нервных клетках мозжечка с вирусными внутриядерными включениями.
Заболевание характеризуется подострым (несколько дней) или постепенным (несколько недель) развитием неврологической и психопатологической симптоматики. Характерно отсутствие общеинфекционных, общемозговых и менингеальных симптомов. Наиболее часто при дебюте заболевания появляются двигательные нарушения (гемипарезы, мозжечковая атаксия), нарушения зрения (гемианопсии), нарушения высших корковых функций (афазия), психические расстройства.
В конечной стадии заболевания наблюдаются глубокая деменция, кома и гибель больного. Течение вариабельно, летальный исход наступает в течение 6-12 мес.
Клиническая картина заболевания, появление и прогрессирование неврологической и психической симптоматики у иммунодефицитного больного заставляют заподозрить ПМЛ. Наибольшие диагностические трудности возникают при СПИДе, когда клиника и МРТ-признаки сходны при ПМЛ и ВИЧ-ассоциированной энцефалопатии [17].
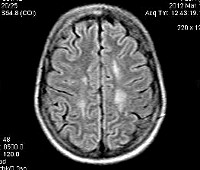
МРТ головного мозга является необходимым методом диагностики при подозрении на ПМЛ. МРТ-картина при подозрении на ПМЛ характеризуется мультифокальными очагами высокой интенсивности сигнала с нечеткими границами на Т2-взвешенных изображениях в подкорковом белом веществе. Чаще поражается белое вещество теменных и затылочных долей, однако очаги могут наблюдаться в любых отдела мозга, включая мозжечок и ствол [1].
В начале заболевания выявляется несколько очагов, по мере прогрессирования процесса отмечается нарастание количества сливных очагов. Очень редко наблюдается незначительный масс-эффект, и в этих случаях их трудно отличить от глиомы. Контрастное усиление отсутствует в результате малой выраженности воспаления. Однако у 5-15% больных отмечается контрастирование по периферии очагов. У 50% больных поражается также и серое вещество [6]. Задняя черепная ямка поражается у 48% больных. Спинной мозг вовлекается крайне редко. Поражение зрительных нервов при ПМЛ не наблюдается.
JC-вирус не вызывает общевоспалительной реакции, в связи с чем изменения в крови и ЦСЖ неспецифичны и не коррелируют с наличием в ЦСЖ JC-вируса. Состав ЦСЖ при ПМЛ у 71% больных не отличается от нормы. У 29% имеется легкое повышение белка (40-80 мг/мл), у 6% - небольшой плейоцитоз (до 16 клеток в 1 мл).
Диагностика ПМЛ в настоящее время основывается на клинических проявлениях заболевания, данных МРТ и результатах исследования ЦСЖ и мозга больных на наличие JC-вируса. Согласно современной классификации, вероятный диагноз может быть выставлен при наличии характерных клинических и нейровизуализационных проявлений при отсутствии JC-вируса в ЦСЖ и мозговой ткани. Лабораторно подтвержденный диагноз устанавливается при наличии ДНК JC-вируса в ЦСЖ больного по данным ПЦР. Гистологически подтвержденный диагноз ПМЛ устанавливается в случае определения JC-вируса методом ПЦР в биопсийном материале мозга больного.
Дифференциальный диагноз ПМЛ должен проводиться со СПИД-деменцией или ВИЧ-энцефалопатией, с оппортунистическими инфекциями ЦНС (энцефалиты цитомегаловирусной, токсоплазменной и грибковой этиологии), а также лимфомой головного мозга. Наибольшие трудности вызывает дифференциальная диагностика с ВИЧ-энцефалопатией, которая может не отличаться ни по клиническим, ни по нейровизуализационным признакам от ПМЛ. В данных случаях только выявление JC-вируса в ЦСЖ и биоптатах головного мозга позволяет установить диагноз [18].
Развитие ПМЛ у больных с аутоиммунными заболеваниями и рассеянным склерозом, получавших терапию моноклональными антителами (ритуксимаб, натализумаб), делает необходимым учитывать возможность развития ПМЛ у этих больных.
Появление нехарактерных клинических симптомов (когнитивные нарушения, афазия, гемианопсия, психические расстройства), прогрессирующее течение ранее ремиттирующего заболевания, появление новых очагов на МРТ, не накапливающих контрастное вещество, являются основаниями предположить наличие ПМЛ у этих пациентов. Основным подтверждением является наличие JC-вируса в ЦСЖ. При отрицательном результате необходимо повторять исследование ЦСЖ каждые 4 нед. Биопсия головного мозга проводится в редких случаях при отсутствии JC-вируса в ЦСЖ при повторных исследованиях.
Дифференциальный диагноз должен проводиться также с токсической лейкоэнцефалопатией, вызванной действием цитостатиков, и различными инфекциями ЦНС (herpes simplex, вирус CMV, varicella zoster, criptocoсcus, aspergillus). В отличие от вышеназванных инфекций, при ПМЛ не встречается общеинфекционных и менингеальных симптомов в связи с отсутствием воспалительной реакции как в ЦНС, так и на периферии.
Исследования последних лет показали, что использование специфических моноклональных антител при рассеянном склерозе, аутоиммунных и лимфопролиферативных заболеваниях повышает риск развития ПМЛ среди пациентов, которые получают эти препараты. К моноклональным антителам относятся такие препараты, как ритуксимаб, натализумаб, инфликсимаб, этанерцепт и др. Ритуксимаб является препаратом моноклональных антител против CD20 предшественников В-лимфоцитов и зрелых В-лимфоцитов. Разрешен к применению при клеточной неходжкинской лимфоме, резистентном ревматоидном артрите. В 2006 г. одобрен FDA для лечения системной краской волчанки. В настоящее время для контроля за действием ритуксимаба и выявления побочных эффектов препарата действует специальный проект (RADAR Research on Advers Drag Events and Report) при участии вирусологов, онкологов, неврологов и других специалистов [3]. Риск развития ПМЛ у больных, получающих ритуксимаб, составляет 1:8000 [16].
На сегодняшний день отмечено 270 случаев развития ПМЛ при применении ритуксимаба; смертность среди этих больных составляет 90%.
Натализумаб - препарат моноклональных антител к α-4 субъединице α-4β1 и α-4β7 интегринов, экспрессирующихся лейкоцитами и являющихся молекулами адгезии; используется в клинической практике с 1999 г. при рассеянном склерозе, ревматоидном артрите, болезни Крона. В связи с развитием 3 случаев ПМЛ у 2 больных рассеянным склерозом и 1 больного болезнью Крона с 28 февраля 2005 г. было приостановлено его применение.
Предполагают несколько механизмов развития ПМЛ при лечении натализумабом: снижение иммунного лейкоцитарного ответа и реактивация JC-вируса; стимуляция высвобождения JC-вируса из костного мозга и незрелых лейкоцитов [14, 26]. Была разработана и утверждена специальная программа для уменьшения риска развития ПМЛ у больных, получающих натализумаб (Risk Minimization Action Plan - Risk MAP). В связи с этим в США действует программа Tysabri Outreach Unified Committee to Health - TOUCH [15, 28].
К февралю 2012 г. умерли 44 (21%) пациента. Большинство смертельных исходов отмечено через 2-3 мес после выявления ПМЛ.
В апреле 2012 г. был описан 1 случай ПМЛ у больного РС, получающего финголимод и в анамнезе терапию натализумабом [9].
Предшествующее назначение иммуносупрессивной терапии, наличие повышенного титра антител к JC-вирусу повышают риск развития ПМЛ у пациентов, получающих иммуносупрессивную терапию длительностью более 24 мес.
В настоящее время разработаны критерии включения и исключения при применении натализумаба у больных с РС [5, 12]. Индивидуальных критериев назначения моноклональных антител после той или иной терапии не разработано [13].
На сегодняшний день эффективного лечения ПМЛ не существует [8]. Используют различные классы лекарственных препаратов: противовирусные средства, цитостатики, антагонисты серотониновых рецепторов, а также трансплантацию стволовых клеток костного мозга [19, 25].
ВААРТ у ВИЧ-инфицированных больных - это мультикомплексная терапия, состоящая из нуклеозидного ингибитора обратной транскриптазы (тимозид, зидовудин), ненуклеозидного ингибитора обратной транскриптазы (делавердин, рескриптор и др.), ингибитора протеаз (санвиновир, инвираза и др.). Лечение ВИЧ и ПМЛ с использованием ВААРТ увеличило выживаемость больных с 3-6 мес до 19,6 мес, снизило заболеваемость ПМЛ и другими оппортунистическими инфекциями ВИЧ-инфицированных больных. В то же время у некоторых больных ВААРТ приводит к манифестации ПМЛ или ухудшению течения ПМЛ, что обусловлено развитием так называемого иммунного реконструктивного воспалительного синдрома (IRIS - immune reconstrictution inflammatory syndrome). Предполагают, что активация инфекции связана с изменением баланса CD8+/CD4+ Т-клеток [20].
Наиболее часто при ПМЛ у не ВИЧ-инфицированных больных используют противовирусные препараты цидофир (вистид), интерферон-альфа, интерлейкин-2.
Стабилизация процесса отмечалась у нескольких больных ПМЛ при лечении цитарабином (известном также как цитозар - ингибитор ДНК полимеразы и репликации вируса).
Современным направлением в лечении ПМЛ является использование препаратов блокаторов 5-гидрокситриптамина-2а серотониновых рецепторов, необходимых для проникновения JC-вируса в клетку.
Оказались способны блокировать JC-вирусную репликацию в олигодендроцитах в ЦНС атипичные антипсихотические средста (рисперидон, оланзапин, зипразидон), которые не только могут приводить к регрессу когнитивных нарушений, но и повышать выживаемость больных.
Наиболее эффективным терапевтическим подходом является восстановление клеточного иммунитета у больных в результате либо снижения дозы иммуносупрессоров, либо их отмены (за исключением состояний после трансплантации органов). Подтверждением этому являются единичные случаи регресса симптоматики и выздоровления больных после отмены цитостатиков [7, 24].
Использование плазмафереза у больных РС, получающих натализумаб, позволило снизить смертность до 21%. По существующим рекомендациям, проведение 5 процедур плазмафереза в течение
10 дней приводит к быстрому восстановлению иммунокомпетентности в головном мозге и способствует своевременной стабилизации состояния пациентов с ПМЛ [28].
Лейкодистрофия — нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
МКБ-10
Общие сведения
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр.), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
Причины возникновения лейкодистрофии
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину — метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток и т. п.
Симптомы лейкодистрофии
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Виды лейкодистрофии
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно-клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван-Богарта-Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Лейкодистрофия Галлервордена-Шпатца чаще всего стартует в 10-летнем возрасте. Проявляется дисфункцией стриопаллидарной системы, затем на фоне гиперкинезов прогрессирует тетрапарез, развивается гемералопия и пигментный ретинит, наблюдается снижение интеллекта, возникают эпиприступы.
Диагностика лейкодистрофии
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха — отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) — возможность пренатального диагностирования.
Лечение лейкодистрофии
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение — в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес.), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Лейкоэнцефалит Шильдера — дегенеративно-демиелинизирующее поражение головного мозга, сопровождающееся образованием крупных или сливных зон демиелинизации. Имеет неуклонно прогрессирующее течение с неспецифичной и полиморфной клинической картиной, которая может включать психические нарушения, пирамидный и экстрапирамидный синдромы, когнитивный дефицит, поражение черепно-мозговых нервов, эписиндром. Диагностируется лейкоэнцефалит Шильдера по клиническим критериям и результатам МРТ после исключения другой патологии с подобными проявлениями. Терапия осуществляется глюкокортикостероидами, антиконвульсантами, миорелаксантами и психотропными средствами. Однако лечение малоэффективно.
МКБ-10
Общие сведения
Преимущественный возраст манифестации болезни Шильдера до сих пор остается спорным вопросом. Зарубежные специалисты в области неврологии считают характерным дебют в возрастном периоде от 7 до 12 лет, а отдельные авторы предлагают относить заболевание к детской форме рассеянного склероза. Наблюдения отечественных неврологов, напротив, свидетельствуют о равной степени поражения лиц различной возрастной категории.
Причины лейкоэнцефалита Шильдера
Этиопатогенез болезни Шильдера находится в стадии изучения. Из названия заболевания видно, что первоначально подразумевалась воспалительная этиология церебрального поражения, т. е. энцефалит. Предполагается вирусная теория заболевания по типу медленных инфекций. Среди возможных инфекционных агентов дискутируется роль кори, герпетической инфекции, миксовирусов, которые, возможно, запускают процесс аутоиммунного церебрального воспаления. Однако безуспешные попытки выделения возбудителя привели к возникновению иной этиопатогенетической теории. Последняя предполагает связь лейкоэнцефалита Шильдера с дисфункцией регуляторных механизмов липидного обмена, что сближает заболевание с наследственными лейкодистрофиями.
Морфологические изменения заключаются в образовании в белом церебральном веществе полушарий значительных зон демиелинизации, имеющих четкие заостренные очертания и зачастую асимметрично расположенных. В ряде случаев подобные очаги формируются в мозжечке и мозговом стволе. У пациентов, заболевших в пубертатном периоде и во взрослом возрасте, описаны случаи, когда наряду с зонами обширной демиелинизации наблюдаются округлые бляшковидные очаги, напоминающие бляшки рассеянного склероза.
Симптомы лейкоэнцефалита Шильдера
Заболевание отличается наличием неспецифичной и полиморфной симптоматики. Может манифестировать исподволь развивающимися психическими расстройствами: лабильностью настроения, апатией, нарушением поведения, эпизодами возбуждения с галлюцинаторным синдромом. Интеллектуальное снижение прогрессирует вплоть до деменции. Наблюдаются аграфия, акалькулия, алексия, агнозия, апраксия. Вследствие демиелинизации черепных нервов возникает неврит зрительного нерва, офтальмоплегия, тугоухость, снижение зрения, бульбарные расстройства. При поражении мозжечка появляется мозжечковая атаксия, скандированная речь, интенционный тремор. Поражение зрительной зоны коры проводит к гемианопсии, корковому амаврозу. Возможны экстрапирамидные нарушения в виде гиперкинезов, торсионной дистонии и т. п. Пирамидные расстройства обычно наблюдаются на поздних этапах лейкоэнцефалита в виде моно-, геми- и тетрапарезов. Зачастую присутствует судорожный синдром (по типу джексоновкой эпилепсии или с генерализованными эпиприступами), характеризующийся отсутствием специфической ЭЭГ-картины.
Вариативность сочетаний различных симптомокомплексов настолько выражена, что не позволяет выделить типичный вариант течения болезни Шильдера. В ряде случаев клиника сходна с прогредиентным вариантом рассеянного склероза, в других - имеет псевдотуморозный характер, в третьих — напоминает психиатрическую патологию. В последнем случае пациенты могут проходить лечение у психиатра вплоть до развития явной неврологической симптоматики.
Диагностика лейкоэнцефалита Шильдера
Прижизненно диагностировать лейкоэнцефалит Шильдера весьма затруднительно. Эта задача требует от невролога тщательного сопоставления анамнестических, клинических и томографических данных, внимательного проведения дифдиагностики со схожими заболеваниями. С целью обследования зрительного и слухового анализаторов к консультациям могут привлекаться офтальмолог и отоларинголог.
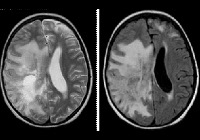
Электроэнцефалография выявляет признаки диффузного церебрального поражения: снижение альфа-активности и дезорганизацию ритма; зачастую определяется эпилептиформная активность. При исследовании цереброспинальной жидкости обнаруживается повышение уровня гамма-глобулина на фоне снижения удельного веса альбуминовой фракции. Наиболее информативным способом инструментальной диагностики выступает МРТ головного мозга. Болезнь Шильдера подтверждает наличие как минимум одного большого или пары сливных очагов демиелинизации в белом церебральном веществе.
Для установления окончательного диагноза многие неврологи руководствуются критериями C.M. Poser 1985 г.: наличие по данным МРТ 1-2 округлых зон демиелинизации величиной не менее 2х3 см; отсутствие патологии надпочечников; исключение любой иной церебральной патологии (внутримозговой опухоли, рассеянного энцефаломиелита, инсульта и пр.); соответствие норме уровня жирных кислот в сыворотке крови; выявление на аутопсии зон диффузного хронического склероза. В некоторых случаях отличить лейкоэнцефалит Шильдера от лейкодистрофии позволяют лишь гистологические исследования церебральных тканей пораженной зоны.
Лечение и прогноз лейкоэнцефалита Шильдера
Отсутствие ясных представлений об этиопатогенезе болезни Шильдера пока не позволило разработать более или менее эффективные методы ее лечения. Отмечен некоторый эффект глюкокортикостероидной терапии, в связи с чем многим пациентам назначают метилпреднизолон, вначале парентерально в ударной дозе, а затем внутрь с постепенным снижением дозы. Параллельно проводится курс нейропротекторной, антиоксидантной и сосудистой терапии, при необходимости назначаются антиконвульсантное лечение (карбамазепин, диазепам), миорелаксанты (амантадин, толперизон, амидин), противоотечные мероприятия (фуросемид, ацетазоламид, магния сульфат), психотропные фармпрепараты.
Своевременно начатое лечение способно лишь несколько задержать прогрессирование патологии. Однако, не смотря на его проведение, все пациенты погибают. Время наступления летального исхода варьирует от нескольких месяцев до 3 лет с момента дебюта лейкоэнцефалита.
Прогрессирующая мультифокальная лейкоэнцефалопатия — редкое демиелинизирующее заболевание, обусловленное реактивацией находящегося в организме большинства людей вируса JC. Патология возникает на фоне угнетения иммунитета у больных СПИДом, гемобластозами, наследственными иммунодефицитами, у пациентов, получающих иммуносупрессивную терапию. Диагностика базируется на клинических данных, результатах томографии головного мозга, ПЦР-исследования ликвора на вирусную ДНК, гистологии церебральных биоптатов. Специфическая терапия не разработана.
Общие сведения
Прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ) ассоциирована с JC-вирусом (JCV), возникает у иммунокомпрометированных пациентов, 85% из которых составляют ВИЧ-инфицированные. Заболевание относится к оппортунистическим инфекциям, носителями вируса являются 90% человечества. До 90-х годов ХХ века заболеваемость ПМЛ не превышала 1 случая на 100 тыс. населения. С ростом числа больных СПИДом этот показатель увеличился до 1 на 20 тыс. человек. Сегодня прогрессирующая лейкоэнцефалопатия наблюдается у 5% больных СПИДом. Некоторые авторы сообщают о снижении заболеваемости за последнее десятилетие в связи с успешным применением антиретровирусной терапии. Одновременно отмечается увеличение распространённости ПМЛ среди лиц с аутоиммунными заболеваниями, что обусловлено использованием в их лечении агрессивной иммунотерапии.
Причины ПМЛ
Прогрессирующая мультифокальная лейкоэнцефалопатия развивается в результате реактивации полиомавируса JC. Вирус распространён повсеместно. Источником инфекции является человек, заражение происходит воздушно-капельным, алиментарным путём. Подавляющее большинство людей заражаются в детстве, являются здоровыми носителями. В течение жизни вирус находится в латентном состоянии, персистирует в почках, селезёнке, костном мозге. Реактивация возбудителя происходит на фоне резко сниженного иммунитета. В группу риска развития заболевания входят следующие состояния:
- ВИЧ-инфецирование. Протекающая в виде СПИДа ВИЧ-инфекция сопровождается угнетением клеточного иммунитета. Выступает самой частой причиной ПМЛ.
- Гемобластозы. Миелопролиферативные (лейкемия) и лимфопролиферативные (лимфомы) процессы приводят к развитию иммунодефицита.
- Аутоиммунная патология: системная красная волчанка, склеродермия, ревматоидный артрит. Иммунодефицит формируется на фоне активного иммуносупрессивного лечения, особенно препаратами моноклональных антител.
- Наследственные заболевания с иммунодефицитом: синдром Ди Джорджи, Вискотта-Олдрича, атаксия-телеангиэктазия.
- Иммуносупрессия на фоне трансплантации органов.
- Вторичный иммунодефицит в результате цитостатической терапии при онкологических заболеваниях.
Патогенез
Расстройство клеточного иммунитета провоцирует перестройку последовательности ДНК JC-вируса, приводит к его активации. Вирус обладает тропностью к клеточным элементам нейроглии (олигодендроцитам, астроцитам), поражение которых сопровождается разрушением миелина. В результате в веществе головного мозга происходит мультифокальная прогрессирующая демиелинизация с ростом и слиянием очагов поражения. Микроскопически обнаруживается увеличение астроцитов, деформация их ядер, окрашивание олигодендроцитов выявляет ядерные включения — скопления частиц JCV. Первостепенную роль в иммунной антивирусной реакции играют цитотоксические Т-лимфоциты, убивающие инфицированные активным вирусом клетки. Снижение выработки специфических Т-лимфоцитов вследствие иммунодефицита обуславливает развитие ПМЛ.
Симптомы ПМЛ
Дебют заболевания носит подострый (2-3 дня) или постепенный (1-3 недели) характер. На первый план выходит патопсихологическая симптоматика и очаговый неврологический дефицит. В типичном варианте прогрессирующая мультифокальная лейкоэнцефалопатия протекает без свойственных нейроинфекциям общемозговых симптомов, менингеального синдрома. Отмечается изменение поведения, агрессивность, эмоциональная лабильность, подозрительность, прогрессирующее ослабление когнитивной сферы (памяти, мышления, внимания). Очаговый дефицит представлен мышечной слабостью конечностей одной половины тела (гемипарезом), афазией, гемианопсией, атаксией, парестезиями в паретичных конечностях. Вначале гемипарез может отсутствовать, в дальнейшем наблюдается у 75% больных. 20% случаев протекают с пароксизмами эпилепсии. Психические расстройства отмечаются у 38% пациентов. Прогрессирование когнитивного дефицита приводит к деменции.
В редких случаях мультифокальная лейкоэнцефалопатия протекает в атипичной форме. К атипичным вариантам относятся JC-менингоэнцефалит, JC-энцефалопатия, гранулярно-клеточная невропатия. Менингоэнцефалитическая форма характеризуется наличием менингеальных симптомов. При JC-энцефалопатии отсутствует очаговый неврологический дефицит. Клиника гранулярно-клеточного варианта представлена изолированным мозжечковым синдромом.
Диагностика
Прогрессирующая лейкоэнцефалопатия диагностируется специалистами в области неврологии на основании клинических данных, результатов нейровизуализирующего исследования, обнаружения специфической ДНК. Диагностический алгоритм включает:
- Осмотр невролога. В классическом варианте в неврологическом статусе определяется гемипарез, гемигипестезия, шаткость, неустойчивость в позе Ромберга, дискоординация, сенсомоторная афазия, когнитивные нарушения. Наблюдается лабильность психики, психопатологические симптомы, возможно неадекватное поведение.
- Осмотр офтальмолога. У большинства пациентов диагностируют снижение зрения, периметрия выявляет гомонимную гемианопсию.
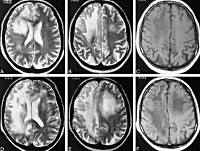
- МРТ головного мозга. Обнаруживается диффузная мультифокальная демиелинизация, очаги имеют различный размер, асимметрично располагаются в белом веществе, таламусе, базальных ядрах.
- ПЦР-исследование. Направлено на выявление ДНК вируса JC в цереброспинальной жидкости, полученной путём люмбальной пункции. Специфичность анализа 90-100%, чувствительность — 70-90%. Проведение антиретровирусной терапии больным СПИДом понижает чувствительность исследования до 58%, отрицательный результат не исключает наличие заболевания.
- Биопсию мозговых тканей. Инвазивная методика, проводится в диагностически затруднительных случаях. Гистологическое исследование образцов церебральных тканей позволяет подтвердить специфические для лейкоэнцефалопатии морфологические изменения.
Лечение ПМЛ
В настоящее время не существует препаратов для лечения прогрессирующей лейкоэнцефалопатии с доказанной эффективностью. Специфическая терапия находится в стадии разработки. Попытки лечения интерфероном, иммуностимуляторами, цитарабином, их комбинациями оказались безрезультатными. Окончились неудачей клинические испытания препарата цидофовир, показывающего анти-JC эффективность на опытах с мышами. Недавно был предложен кардинально новый метод лечения антидепрессантом миртазапином, блокирующим распространение JCV благодаря связыванию рецепторов, через которые вирус инфицирует клетки нейроглии. Способ требует клинических испытаний.
Прогноз и профилактика
Прогрессирующая мультифокальная лейкоэнцефалопатия отличается неуклонно усугубляющимся течением с исходом в кому. Продолжительность жизни варьирует от 1 мес. (острая форма) до 10-12 мес. с момента заболевания. Профилактика подразумевает меры предупреждения инфицирования ВИЧ, осторожное проведение терапии аутоиммунных заболеваний, мониторинг неврологической симптоматики у больных, получающих лечение моноклональными препаратами.
Лейкоэнцефалопатия — хроническая прогрессирующая патология, обусловленная деструкцией белого вещества головного мозга и приводящая к старческой деменции или слабоумию. Заболевание имеет несколько равнозначных названий: энцефалопатия Бинсвангера или болезнь Бинсвангера. Автор впервые описал патологию в 1894 году и дал ей свое имя. Наряду с сосудистой лейкоэнцефалопатией, в последние годы свое распространение получает прогрессивная мультифокальная лейкоэнцефалопатия (ПМЛ) – заболевание вирусной этиологии.
Гибель нервных клеток, вызванная нарушением кровоснабжения и гипоксией мозга, приводит к развитию микроангиопатии. Лейкоареоз и лакунарные инфаркты изменяют плотность белого вещества и указывают на имеющиеся в организме проблемы с циркуляцией крови.
Клиника лейкоэнцефалопатии зависит от степени тяжести и проявляется различной симптоматикой. Обычно признаки подкорковой и лобной дисфункции сочетаются с эпиприпадками. Течение патологии хроническое, характеризующееся частой сменой периода стабилизации и обострения. Лейкоэнцефалопатия встречается преимущественно у пожилых людей. Прогноз заболевания неблагоприятный: быстро развивается тяжелая инвалидизация.
Формы лейкоэнцефалопатии
Мелкоочаговая лейкоэнцефалопатия
Мелкоочаговая лейкоэнцефалопатия — хроническое заболевание сосудистого происхождения, основной причиной которого является гипертония. Стойкая гипертензия приводит к постепенному поражению белого вещества мозга.
В наибольшей степени развитию лейкоэнцефалопатии сосудистого генеза подвержены мужчины в возрасте 55 лет и старше с наследственной предрасположенностью. Сосудистая лейкоэнцефалопатия – хроническая патология сосудов головного мозга, приводящая к поражению белого вещества и развивающаяся на фоне стойкой гипертензии.
О сосудистых энцефалопатиях рекомендуем подробную информацию по ссылке.
Прогрессирующая мультифокальная лейкоэнцефалопатия
Прогрессирующая мультифокальная энцефалопатия – вирусное поражение ЦНС, приводящее у иммунологически скомпрометированных лиц к разрушению белого вещества. Вирусы еще больше подавляют иммунную защиту, развивается иммунодефицит.

поражение белого вещества мозга при лейкоэнцефалопатии
Эта форма патологии является самой опасной и часто заканчивается смертью больного. Но с созданием и совершенствованием антиретровирусной терапии распространенность заболевания уменьшились в несколько раз.
Прогрессирующая мультифокальная лейкоэнцефалопатия поражает больных врожденным или приобретенным иммунодефицитом. Патологию обнаруживают у 5 % ВИЧ-инфицированных больных и у 50% больных СПИДом.
Симптоматика заболевания многообразна. Когнитивные нарушения варьируются от легкой дисфункции до выраженной деменции. Очаговая неврологическая симптоматика характеризуется нарушением речи и зрения вплоть до слепоты, а отдельные двигательные расстройства быстро прогрессируют и часто приводят к тяжелой инвалидности.
Перивентрикулярная лейкоэнцефалопатия
Перивентрикулярная форма – поражение подкорковых мозговых структур, возникающее на фоне хронической гипоксии и острой сосудистой недостаточности. Очаги ишемии беспорядочно разбросаны в структурах нервной системы и в основном веществе мозга. Заболевание начинается с поражения двигательных ядер продолговатого мозга.
Лейкоэнцефалопатия с исчезающим белым веществом
Лейкоэнцефалопатия с исчезающим белым веществом — генетически обусловленная патология, причиной которой является мутация в генах. Классическая форма заболевания впервые проявляется у детей 2 – 6 лет.
У больных прогрессируют: мозжечковая атаксия, тетрапарезы, мышечная недостаточность, когнитивные расстройства, оптическая атрофия, эпиприступы. У младенцев нарушается процесс вскармливания, возникает рвота, лихорадка, задерживается психомоторное развитие, повышается возбудимость, развивается гипертонус конечностей, судорожный синдром, ночная задержка дыхания, кома.
Причины
В большинстве случаев лейкоэнцефалопатия – следствие стойкой гипертонии. Пациенты – пожилые люди, с сопутствующим атеросклерозом и ангиопатиями.
Другие заболевания, осложняющиеся возникновением лейкоэнцефалопатии:
- Синдром приобретенного иммунодефицита,
- Лейкоз и другие онкологические заболевания крови,
- Лимфогранулематоз,
- Туберкулез легких,
- Саркаидоз,
- Раковые опухоли внутренних органов,
- Длительный прием иммунодепрессантов также провоцирует развитие данной патологии.
Рассмотрим развитие поражения мозга на примере прогрессирующей мультифокальной энцефалопатии.
Вирусы, ставшие причиной ПМЛ тропны к нервных клеткам. Они содержат двухцепочечную кольцевую ДНК и избирательно поражают астроциты и олигодендроциты, синтезирующие миелиновые волокна. В ЦНС появляются очаги демиелинизации, нервные клетки увеличиваются и деформируются. Серое вещество мозга в патологию не вовлекается и остается абсолютно незатронутым. Белое вещество изменяет свою структуру, становится мягким и студенистым, на нем появляются многочисленные маленькие впадины. Олигодендроциты становятся пенистыми, астроциты приобретают неправильную форму.

поражение мозга при прогрессирующей мультифокальной лейкоэнцефалопатии
Полиомавирусы – мелкие микробы, лишенные суперкапсида. Они являются онкогенными, находятся в организме хозяина длительно в латентном состоянии и не вызывают болезнь. При снижении иммунной защиты эти микробы становятся возбудителями смертельно опасного заболевания. Выделение вирусов — сложнейшая процедура, которая проводится только в специализированных лабораториях. С помощью электронной микроскопии в срезах олигодендроцитов вирусологи обнаруживают вирионы полиомавирусов, имеющих форму кристалла.
Полиомавирусы проникают в организм человека и находятся в латентном состоянии во внутренних органах и тканях пожизненно. Персистенция вирусов происходит в почках, костном мозге, селезенке. При снижении иммунной защиты они активизируются и проявляют свое патогенное действие. Они транспортируются лейкоцитами в ЦНС и размножаются в белом веществе головного мозга. Подобные процессы происходят у лиц, страдающих СПИДом, лейкемией или лимфомой, а также перенесших трансплантацию органов. Источником инфекции является больной человек. Вирусы могут передаваться воздушно-капельным или фекально-оральным путем.
Симптоматика
Заболевание развивается постепенно. Сначала больные становятся неловкими, рассеянными, апатичными, слезливыми и неуклюжими, у них снижается умственная работоспособность, нарушается сон и память, затем появляется вялость, общая утомляемость, вязкость мысли, шум в ушах, раздражительность, нистагм, мышечный гипертонус, сужается круг интересов, с трудом произносятся некоторые слова. В запущенных случаях возникают моно- и гемипарезы, неврозы и психозы, поперечный миелит, судороги, нарушение высших мозговых функций, грубая деменция.

Ведущими симптомами болезни являются:
- Дискоординая движений, шаткость походки, двигательная дисфункция, слабость в конечностях,
- Полный односторонний паралич рук и ног,
- Нарушения речи,
- Снижение резкости зрения,
- Скотомы,
- Гипестезия,
- Снижение интеллекта, спутанность сознания, эмоциональная лабильность, слабоумие,
- Гемианопсия,
- Дисфагия,
- Эпиприступы,
- Недержание мочи.
Прогрессирующая мультифокальная лейкоэнцефалопатия проявляется вялыми парезами и параличами, типичной гомонимной гемианопсией, оглушенностью, изменением личности, симптомами поражения черепно-мозговых нервов и экстрапирамидными нарушениями.
Диагностика
Диагностика лейкоэнцефалопатии включает проведение целого ряда процедур:
- Консультацию у врача-невропатолога,
- Клинического анализа крови,
- Выявления уровня алкоголя, кокаина и амфетамина в крови,
- Допплерографию,
- ЭЭГ,
- КТ, МРТ,
- Биопсию головного мозга,
- ПЦР,
- Люмбальную пункцию.
С помощью КТ и МРТ можно обнаружить гиперинтенсивные очаги поражения в белом веществе мозга. При подозрение на инфекционную форму, электронная микроскопия позволяет выявить в мозговой ткани частицы вирусов. Иммуноцитохимический метод – выявление антигена вируса. Люмбальную пункцию проводят при повышении белка в СМЖ. При данной патологии в ней выявляют также лимфоцитарный плеоцитоз.
Подтвердить или опровергнуть диагноз лейкоэнцефалопатия могут результаты тестов на психологическое состояние, память, координацию движений.
Лечение
Лечение лейкоэнцефалопатии длительное, комплексное, индивидуальное, требующее от больного много сил и терпения.
Лейкоэнцефалопатия — заболевание неизлечимое. Общетерапевтические мероприятия направлены на сдерживание дальнейшего прогрессирования патологии и восстановление функций подкорковых структур головного мозга. Лечение лейкоэнцефалопатии симптоматическое и этиотропное.
Дополнительно проводят физиотерапию, рефлексотерапию, назначают дыхательную гимнастику, массаж воротниковой зоны, мануальную терапию, иглоукалывание. Для лечения детей медикаменты обычно заменяют гомеопатическими и фитотерапевтическими препаратами.
Лейкоэнцефалопатия, наряду со старческой формой с прогрессирующей деменцией, в последнее время стала осложнением СПИДа, что связано с сильно ослабленным иммунитетом ВИЧ-инфицированных пациентов. При отсутствии своевременной и адекватной терапии такие пациенты живут не 6 месяцев с момента появления клинических симптомов патологии. Лейкоэнцефалопатия всегда заканчивается смертью больного.
Читайте также: