
Что такое медленная вирусная инфекция
Обновлено: 25.04.2024
группа вирусных заболеваний человека и животных, характеризующихся продолжительным инкубационным периодом, своеобразием поражений органов и тканей, медленным течением со смертельным исходом.
На основании сделанных открытий первоначально возникло предположение о существовании в природе особой группы медленных вирусов. Однако вскоре была установлена его ошибочность, во-первых, благодаря открытию у ряда вирусов, являющихся возбудителями острых инфекций (например, у вирусов кори, краснухи, лимфоцитарного хориоменингита, герпеса), способности вызывать также медленные вирусные инфекции, во-вторых, — в связи с обнаружением у возбудителя типичной М.в.и. — вируса висны — свойств (структуры, размеры и химический состав вирионов, особенности репродукции в клеточных культурах), характерных для широкого круга известных вирусов.
В соответствии с особенностями этиологических агентов М.в.и. подразделяют на две группы: к первой относятся М.в.и., вызываемые вирионами, ко второй — прионами (инфекционными белками). Прионы состоят из белка с молекулярной массой 27 000—30 000. Отсутствие в составе прионов нуклеиновых кислот определяет необычность некоторых из свойств: устойчивость к действию β-пропиолактона, формальдегида, глутаральдегида, нуклеаз, псораленов, УФ-излучения, ультразвука, ионизирующего излучения, к нагреванию до t° 80° (при неполной инактивации даже в условиях кипячения). Ген, кодирующий прионовый белок, находится не в составе приона, а в клетке. Прионовый белок, попадая в организм, активирует этот ген и вызывает индукцию синтеза аналогичного белка. Вместе с тем прионы (называемые также необычными вирусами) при всем своем структурном и биологическом своеобразии обладают рядом свойств обычных вирусов (вирионов). Они проходят через бактериальные фильтры, не размножаются на искусственных питательных средах, репродуцируются до концентраций 10 5 —10 11 на 1 г мозговой ткани, адаптируются к новому хозяину, изменяют патогенность и вирулентность, воспроизводят феномен интерференции, обладают штаммовыми различиями, способностью к персистенции в культуре клеток, полученных из органов зараженного организма, могут быть клонированы.
Группа М.в.и., вызываемых вирионами, включает около 30 заболеваний человека и животных. Вторая группа объединяет так называемые подострые трансмиссивные губкообразные энцефалопатаи, включающие четыре М.в.и. человека (куру, болезнь Крейтцфельдта — Якоба, синдром Герстманна — Штраусслера, амиотрофический лейкоспонгиоз) и пять М.в.и. животных (скрепи, трансмиссивную энцефалопатию норок, хроническую изнуряющую болезнь находящихся в неволе оленей и лосей, губкообразную энцефалопатию коров). Кроме упомянутых существует группа заболеваний человека, каждое из которых по клиническому симптомокомплексу, характеру течения и исходу соответствует признакам М.в.и., однако причины этих заболеваний точно не установлены и поэтому их причисляют к категории М.в.и. с предполагаемой этиологией. К ним относят вилюйский энцефаломиелит, Рассеянный склероз, Амиотрофический боковой склероз, болезнь Паркинсона (см. Паркинсонизм) и ряд других.
Эпидемиология М.в.и. имеет ряд особенностей, прежде всего связанных с их географическим распространением. Так, куру эндемична для восточного плоскогорья о. Новая Гвинея, а вилюйский энцефаломиелит — для районов Якутии, главным образом примыкающих к р. Вилюй. Рассеянный склероз не известен на экваторе, хотя заболеваемость в северных широтах (то же для южного полушария) достигает 40—50 на 100 000 чел. При повсеместном относительно равномерном распространении амиотрофического бокового склероза заболеваемость на о. Гуам в 100 раз, а на о. Новая Гвинея в 150 раз выше, чем в других частях света.
При врожденной краснухе (Краснуха), синдроме приобретенного иммунодефицита (см. ВИЧ-инфекция), куру, Крейтцфельдта — Якоба болезни (Крейтцфельдта — Якоба болезнь) и др. источником инфекции является больной человек. При прогрессирующей многоочаговой лейкоэнцефалопатии, рассеянном склерозе, болезни Паркинсона, вилюйском энцефаломиелите, амиотрофическом боковом склерозе, рассеянном склерозе источник не известен. При М.в.и. животных источником инфекции служат больные животные. При алеутской болезни норок, лимфоцитарном хориоменингите мышей, инфекционной анемии лошадей, скрепи существует риск заражения людей. Механизмы передачи возбудителей разнообразны и включают контактный, аспирационный и фекально-оральный; возможна также передача через плаценту. Особую эпидемиологическую опасность представляет такая форма течения М.в.и. (например, при скрепи, висне и др.), при которой скрытое вирусоносительство и типичные морфологические изменения в организме протекают бессимптомно.
Патогистологические изменения при М.в.и. можно подразделить на ряд характерных процессов, среди которых прежде всего следует назвать дегенеративные изменения в ц.н.с. (у человека — при куру, болезни Крейтцфельдта — Якоба, амиотрофическом лейкоспонгиозе, амиотрофическом боковом склерозе, болезни Паркинсона, вилюйском энцефаломиелите; у животных — при подострых трансмиссивных губкообразных энцефалопатиях, медленной гриппозной инфекции мышей и др.). Нередко поражения ц.н.с. сопровождаются процессом демиелинизации, особенно ярко выраженным при прогрессирующей многоочаговой лейкоэнцефалопатии. Воспалительные процессы достаточно редки и, например, при подостром склерозирующем панэнцефалите, прогрессирующем краснушном панэнцефалите, висне, алеутской болезни норок носят характер периваскулярных инфильтратов.
Общей патогенетической основой М.в.и. является накопление возбудителя в различных органах и тканях зараженного организма задолго до первых клинических проявлений и длительное, иногда многолетнее, размножение вирусов нередко и в тех органах, в которых никогда не обнаруживают патогистологических изменений. При этом важным патогенетическим механизмом М.в.и. служит цитопролиферативная реакция различных элементов. Так, например, губкообразные энцефалопатии характеризуются выраженным глиозом, патологической пролиферацией и гипертрофией астроцитов, что и влечет за собой вакуолизацию и гибель нейронов, т.е. развитие губкообразного состояния ткани мозга. При алеутской болезни норок, висне и подостром склерозирующем панэнцефалите наблюдается резко выраженная пролиферация элементов лимфоидной ткани. Многие М.в.и., такие как прогрессирующая многоочаговая лейкоэнцефалопатия, лимфоцитарный хориоменингит новорожденных мышей, прогрессирующая врожденная краснуха, медленная гриппозная инфекция мышей, инфекционная анемия лошадей и др., могут быть обусловлены выраженным иммунодепрессирующим действием вирусов, образованием иммунных комплексов вирус — антитело и последующим повреждающим действием этих комплексов на клетки тканей и органов с вовлечением в патологический процесс аутоиммунных реакций.
Ряд вирусов (вирусы кори, краснухи, герпеса, цитомегалии и др.) способны вызывать М.в.и. в результате внутриутробного заражения плода.
Клиническому проявлению М.в.и. иногда (куру, рассеянный склероз, вилюйский энцефа-ломиелит) предшествует период предвестников. Только при вилюйском энцефаломиелите, лимфоцитарном хориоменингите у людей и инфекционной анемии лошадей заболевания начинаются с повышения температуры тела. В большинстве же случаев М.в.и. возникают и развиваются без температурной реакции организма. Все подострые трансмиссивные губкообразные энцефалопатии, прогрессирующая многоочаговая лейкоэнцефалопатия, болезнь Паркинсона, висна и др. проявляются нарушениями походки и координации движений. Нередко эти симптомы оказываются наиболее ранними, позднее к ним присоединяются гемипарезы и параличи. При куру и болезни Паркинсона характерно дрожание конечностей; при висне, прогрессирующей врожденной краснухе — отставание в массе тела и росте. Течение М.в.и., как правило, прогрессирующее, без ремиссий, хотя при рассеянном склерозе и болезни Паркинсона могут наблюдаться ремиссии, увеличивающие продолжительность заболеваний до 10—20 лет.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
История

Гистологический препарат головного мозга, на котором видны микрополости


Как уже было сказано выше, основы знаний о прионах заложил Стенли Прузинер. Немного из его биографии. Родился в США в 1942 году. Его предки - эмигранты из российской империи, еврейского происхождения, вынужденные покинуть страну из-за еврейских погромов. Сам Стенли Прузинер в 1968 г. закончил Университет Пенсильвании и работал ординатором-неврологом в Медицинской школе Калифорнийского университета (Сан-Франциско). В 1970 впервые встретился с болезнью Крейтцфельдта — Якоба. У пациента, находившегося на лечении у Прузинера, никак не выявлялся возбудитель. Плотно занявшись этим исследованием, невролог обратился к трудам другого врача – Сиггурдсона, выявившего определенные закономерности у непонятных на тот момент болезней.
Такими закономерностями стали:
- необычно продолжительный (месяцы и годы) инкубационный период;
- медленно прогрессирующий характер течения;
- необычность поражения органов и тканей;
- неизбежность смертельного исхода.
Что же такое прионы и каков их механизм действия на организм (современные представления)?
На самом деле в организме человека и многих других живых существ есть белки PrPC. По-русски – нормальная форма прионных белков (открыты были после исследований Сиггурдсона, поэтому такая странность в название). Известна его длина, последовательность аминокислот, вторичная структура. Важно знать, что конечная структура состоит из трёх α-спиралей и двухцепочечного антипараллельного β-листа. Обладают интересным свойством, а именно осаждаются высокоскоростным центрифугированием, что является стандартным тестом на наличие прионов. Есть данные, что PrP играет важную роль в прикреплении клеток, передаче внутриклеточных сигналов, а потому может быть вовлечён в коммуникацию клеток мозга. Тем не менее, функции PrP исследованы недостаточно.

(a) норма (b) патология
Считается, что прионное заболевание может быть приобретено 3 путями: в случае прямого заражения, наследственно или спорадически (спонтанно) или их комбинациями. Спорадическая (то есть спонтанная) прионная болезнь возникает в популяции у случайной особи. Таков, например, классический вариант болезни Крейтцфельдта — Якоба. Существуют две основные гипотезы относительно спонтанного появления прионных болезней. Согласно первой из них спонтанное изменение происходит в самом доселе нормальном белке в мозге, то есть имеет место посттрансляционная модификация. Альтернативная гипотеза гласит, что одна или несколько клеток организма в какой-то момент претерпевают соматическую мутацию (то есть, не передающуюся наследственно) и начинают производить дефектный белок PrPSc. Как бы то ни было, конкретный механизм спонтанного возникновения прионных болезней неизвестен. Вторая – заражение. По данным современных исследований, основной путь приобретения прионных заболеваний — поедание заражённой пищи. Считается, что прионы могут оставаться в окружающей среде в останках мёртвых животных, а также присутствуют в моче, слюне и других жидкостях и тканях тела (кровь, ликвор). Из-за этого заражение прионами может произойти и в ходе пользования нестерильными хирургическими инструментами. Это усложняет стерилизацию хирургических инструментов или устройств на скотобойне. Прионы в большинстве своём устойчивы к протеазам, высокой температуре, радиации и хранению в формалине, хотя эти меры и снижают их способность к заражению. Эффективная дезинфекция против прионов должна включать гидролиз или повреждение/разрушение их третичной структуры. Это можно достичь обработкой хлорной известью, гидроксидом натрия и сильнокислыми моющими веществами. Пребывание в течение 18 минут при температуре 134 °C в герметичном паровом автоклаве не может деактивировать прионы. В качестве основного современного метода для деактивации и денатурации прионов в настоящее время изучается озоновая стерилизация. Ренатурация полностью денатурированного приона до инфективного состояния зафиксирована не была, однако для частично денатурированных прионов в некоторых искусственных условиях это возможно. Еще стоит помнить, что эти белки могут долго сохраняться в почве за счёт связывания с глиной и другими почвенными минералами. Не впадайте в паранойю, но теоретически они могут быть повсюду. В 2011 году было сообщено об открытии прионов, передающихся по воздуху в частицах аэрозоля (то есть воздушно-капельным путём). Также в 2011 году было опубликовано предварительное доказательство того, что прионы могут передаваться с получаемым из мочи человеческим менопаузальным гонадотропином, применяемым для лечения бесплодия. Теоретически с помощью всего одного больного животного с прионной болезнью, можно уничтожать целые нации и страны, просто добавляя его костную муку в кормовые добавки и продавая их в нужное государство. Сходная ситуация произошла в конце 80-х годов в Британии (эпидемия коровьего бешенства). Тогда, скорее всего по незнанию (а не по злому умыслу) произошел вышеуказанный процесс, унесший жизни около 200 человек (на 2009 год) и 179 тыс. голов крупного рогатого скота.

Клиника
Поговорим о болезнях и клинических проявлениях. Теоретически может возникать у всех живых существ, обладающих PrPc Вот некоторые примеры. У овец и коз, как это уже говорилось выше, главное проявление - это скрейпи. Для коров характерно коровье бешенство (губчатая энцефалопатия крупного рогатого скота) У норок- Трансмиссивная энцефалопатия норок. И так далее. Зафиксированы проявления заболеваний у кошек, диких парнокопытных, страусов. Но нас интересуют болезни человека.
Болезнь Крейтцфельдта — Якоба. Код по МКБ-10 A81.0; F02.1. Код А соответствует инфекционным болезням (А81 – инфекционные болезни нервной системы). Код F – психические расстройства, F02 – деменции.

Темно зеленый распространение К-Я
Светло зеленый - коровьего бешенства
Основные клинические критерии для постановки диагноза:
Выделяют несколько клинических форм:
Спонтанная — классическая (sCJD) Согласно современным представлениям (прионной теории), прионы при этой форме заболевания возникают в мозге спонтанно, без какой-либо видимой внешней причины. Болезнь обычно поражает людей в возрасте старше 50 лет и проявляется с вероятностью 1-2 случая на миллион жителей. Вначале проявляется в форме кратких потерь памяти, изменениями настроения, потерей интереса к происходящему вокруг. Далее симптомы деменции прогрессируют со всеми вытекающими последствиями.
Наследственная (fCJD) Болезнь возникает в семьях, где наследуется повреждение гена для прионового белка. Дефектный прионовый белок является намного более подверженным спонтанному превращению в прион. Признаки и ход болезни подобны классической форме.
Ятрогенная (1CJD) Болезнь обусловлена непреднамеренным внесением прионов в тело пациента при медицинском вмешательстве. Источником прионов ранее были некоторые лекарства, инструменты или мозговые оболочки, которые забирались у мертвых людей и использовались для закрытия раны при операциях на мозге. Признаки и ход болезни подобны классической форме. Новый вариант (nvCJD) Болезнь появилась впервые в 1995 году в Великобритании и с того момента от нее умерло не более 100 человек. Вероятнее всего, что они заразились мясными продуктами, содержащими бычьи прионы.
- психические расстройства и сенсорные нарушения,
- характерны глобальные когнитивные нарушения и атаксия.
- описано несколько случаев заболевания, дебютировавшего с корковой слепоты (вариант Heidenhain).
- эписиндром представлен также миоклоническими припадками.
- мозжечковая симптоматика выявляется в 100 %.
- Пациент страдает от всё более тяжёлой бессонницы, панических атак и фобий. Эта стадия длится в среднем 4 месяца.
- Панические атаки становятся серьёзной проблемой, и к ним присоединяются галлюцинации. Эта стадия длится в среднем 5 месяцев.
- Полная неспособность спать, сопровождаемая быстрой потерей веса. Эта стадия длится в среднем 3 месяца.
- Пациент перестаёт говорить и не реагирует на окружающее. Это последняя стадия болезни, длящаяся в среднем 6 месяцев, после чего пациент умирает.
Куру, почти не встречается в настоящее время, в связи с искоренением каннибализма. Интересно, что в 2009 году американские учёные сделали неожиданное открытие: некоторые члены племени форе, благодаря появившемуся у них в сравнительно недавнем времени новому полиморфизму гена PRNP, имеют врождённый иммунитет к куру.
В настоящее время нет ни одного средства останавливающего или тормозящего развитие прионных болезней.
Прионы. Прионовые инфекции. Патогенез прионовых инфекций.
Особую группу медленных инфекций составляют поражения ЦНС, проявляющиеся вакуолизацией серого вещества — так называемым губкообразным перерождением нервных тканей. Ему предшествует развитие первично-дегенеративных процессов при полном отсутствии воспалительных реакций.
Своеобразие патоморфологической картины обусловило первичное название заболеваний — трансмиссивные спонгиоформные [от греч. spongia, губка] энцефалопатии.
Было установлено, что возбудители прионовых инфекций проходят через бактериальные фильтры, не размножаются на искусственных питательных средах, воспроизводят феномен интерференции, что дало основание отнести их к вирусам. Однако американский вирусолог Гайдушек установил, что эти вирусы обладают необычными свойствами, так как они оказались устойчивы к действию многих вирулицидных факторов, а материалом, полученным от погибших животных и людей, невозможно заразить клеточные культуры.
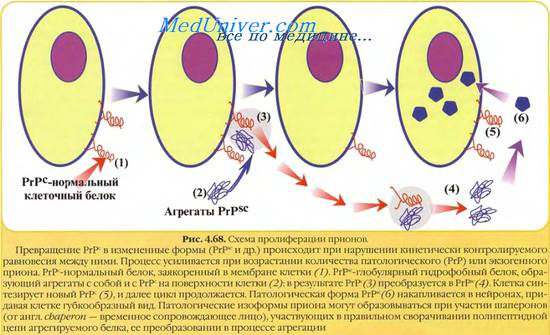
Патогенез поражений прионовых инфекций обусловлен способностью инфекционного прионового белка РrР с (от англ. scrapie, скрепи, являющейся самой распространённой прионовой болезнью) вызывать мутацию гена, кодирующего синтез нормального нейронального прионового белка РrР [от англ. cell, клетка], в результате чего синтезируется инфекционный прионовый белок РrРsс, отличающийся нарушенной пространственной конфигурацией молекулы.
Молекула РrРsс соединяется с молекулой РrРsс с образованием димерного продукта, трансформирующегося в 2 молекулы РrРsс . В следующем цикле 2 молекулы РгР соединяются с 2 молекулами РrРsс, давая начало 4 молекулам PrPSc, что обеспечивает экспоненциальное образование молекул PrPSc. Таким образом, образование инфекционных прионовых белков происходит не за счёт репродукции молекулы РrРsс, попавшей в организм, а за счёт синтеза новых молекул, кодируемых мутировавшим геном РrР .
Физиологическое значение белка РrРsс прионовых инфекций связывают с реализацией функций синапсов, сохранением клеток Пуркинье, регуляцией внутриклеточного содержания Са 2+ в нейронах, поддержанием трофики некоторых их популяций и сохранением резистентности нейронов и астроцитов к повреждающим факторам. Белок PrP — короткоживущий (период полураспада 5-6 ч).
В противоположность этому инфекционный прионовый белок PrPSc накапливается в цитоплаз-менных везикулах, что приводит к последующему нарушению функций синапсов и развитию глубоких неврологических дефектов. Позднее PrP с высвобождается во внеклеточное пространство и откладывается в амилоидных бляшках.
У человека прионы вызывают куру, болезнь Кройтцфельдта-Якоба, синдром Гtрстмана-Штраусслера-Шайнкера и фатальную семейную бессонницу. Их характерная особенность — практически полное отсутствие иммунных реакций к инфекционным прионовым белкам, что связано с их внутриклеточной локализацией и структурным сходством с нейрональными белками. Гистологически в тканях мозга выявляют выраженную губчатую дегенерацию, генерализованную гипертрофию астроцитов и амилоидные бляшки, состоящие из белка PrPSc.
Изучение условий возникновения прионовых болезней выявило уникальные особенности их эпидемиологии, отличающие их от прочих инфекционных заболеваний. Они могут формироваться как инфекционные, спорадические и наследственные поражения. В последнем случае предполагают наличие генетической предрасположенности к прионовым инфекционным белкам.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
На уровне макроорганизма основные формы вирусных поражений принципиально не отличаются от таковых, наблюдаемых при инфицировании вирусами отдельных клеток.
Продуктивная вирусная инфекция с образованием дочерних популяций и характерными клиническими проявлениями возможна лишь при наличии в заражённом организме чувствительных клеток, в которых осуществляется репродуктивный цикл возбудителя. Например, возбудитель полиомиелита может реплицировать только в клетках ЖКТ и ЦНС приматов и человека.
Абортивная инфекция развивается при проникновении возбудителя в нечувствительные клетки (например, при попадании вируса лейкоза коров в организм человека) либо в клетки, не способные обеспечить полный репродуктивный цикл (например, находящиеся в стадии клеточного цикла G0). Способность клеток к поддержанию вирусспецифических репродуктивных процессов также подавляет ИФН, противовирусный эффект которого направлен против самых различных вирусов.

Персистирующая вирусная инфекция возникает при таком взаимодействии между вирусом и заражённой клеткой, когда в последней продолжается выполнение собственных клеточных функций. Если заражённые клетки делятся, образуется инфицированный клон. Таким образом, увеличение числа заражённых клеток способствует увеличению общей популяции возбудителя в организме. Тем не менее персистирующие вирусные инфекции обычно нарушают функции клеток, что в конце концов приводит к клиническим проявлениям. У человека развитие персисти-рующих инфекций в определённой степени зависит от возраста. Например, внутриутробное заражение вирусом коревой краснухи или цитомегаловирусом (ЦМВ) приводит к ограниченному по времени персистированию возбудителя. Появление симптоматики связано с возможностью плода развивать иммунные реакции на инфекционный агент.
Латентная (скрытая) вирусная инфекция. В то время как персистирующие инфекции сопровождаются постоянным высвобождением дочерних вирусных популяций, при латентных поражениях они образуются спорадически. Репродуктивный цикл подобных возбудителей резко замедляется на поздних стадиях и активируется под влиянием различных факторов. Латентные инфекции характерны для большинства герпесвирусов, вызывающих рецидивирующие и обычно не прогрессирующие заболевания.

Дремлющая (криптогенная) вирусная инфекция — форма проявления вирусной инфекции при которой возбудитель в неактивном состоянии находится в отдельных очагах (например, в нервных ганглиях). Клинически инфекция проявляется лишь при резком ослаблении защитных сил организма. Например, вирус герпеса 3 типа, вызывающий при первичном заражении ветряную оспу, пожизненно сохраняется в организме. Рецидив заболевания в форме опоясывающего лишая возможен лишь при нарушениях иммунного статуса (наиболее часто в пожилом возрасте).
Медленные вирусная инфекции характеризуются длительным инкубационным периодом (месяцы и годы), в течение которого возбудитель размножается, вызывая всё более явные повреждения тканей. Первоначально возбудитель размножается в ограниченной группе клеток, но постепенно инфицирует всё большее их число. Заболевания заканчиваются развитием тяжёлых поражений и смертью больного. К медленным вирусным инфекциям относят подострый склерозирующий панэнцефалит, ВИЧ-инфекцию и др.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Читайте также: