
Вирус человеческого т-клеточного лейкоза
Обновлено: 25.04.2024
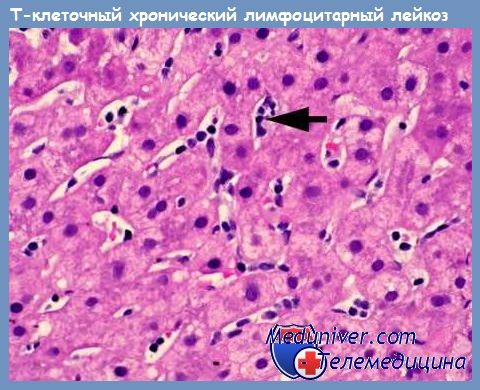
Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) - диагностика, лечение
В середине 70-х годов XX в. одновременно с открытием иммунологических маркеров В- и Т-лимфоцитов появилась возможность разделения этих клеточных популяций. Соответственно двум типам лимфоцитов были выделены две основные группы лимфопролиферативных заболеваний: В- и Т-клеточные. Первый иммунологический тест, позволивший продемонстрировать Т-клеточную природу лимфоцитов, основывался на способности этих клеток формировать розетки при инкубации с эритроцитами барана (Е-розетки).
Совершенствование лабораторных методов исследования, особенно иммунологических и молекулярно-генетических, позволило более детально охарактеризовать и классифицировать эту группу заболеваний. Т-клеточные лимфопролиферативные заболевания составляют 10—15 % от всех опухолей лимфатической системы и по уровню дифференцировки и созревания могут быть разделены на две группы: тимические и посттимические.
Посттимические опухоли представлены иммунологически зрелыми Т-лимфоцитами, в ядрах которых отсутствует фермент терминальная дезоксинуклеотидилтрансфераза (TdT).
Одной из форм зрелоклеточных Т-клеточных лимфопролиферативных заболеваний является Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ), впервые описанный D. Catovsky и соавт. в 1973 г.. Наиболее часто Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) встречается в двух возрастных группах: пожилых (средний возраст 69 лет) и молодых с атаксией-телеангиэктазией (AT). Женщины болеют чаще, чем мужчины, в соотношении 4:1. Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) характеризуется агрессивным течением с медианой выживаемости 7,5 мес.
Заболевание начинается остро или подостро. Больные жалуются на быструю утомляемость, слабость, потливость, снижение массы тела. Одними из первых симптомов могут быть боли в животе, связанные с выраженной спленомегалией, увеличением внутри-брюшных лимфатических узлов, а также гематологические изменения (анемия, тромбоцитопения), обусловленные костно-мозговой недостаточностью и гиперспленизмом. Реже первой манифестацией заболевания является поражение кожи, отличающееся полиморфной картиной, — от кожной сыпи, обычно пятнисто-папулезной, до генерализованной эритродермии.
Органные поражения, например ЦНС и легких, встречаются редко. Менее чем у 5 % больных заболевание начинается бессимптомно, и только в анализе крови обнаруживается медленно нарастающий абсолютный лимфоцитоз. Такие случаи, особенно мелкоклеточный вариант Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ), ошибочно диагностируют как хронический лимфолейкоз (ХЛЛ). В противоположность ХЛЛ, течение которого может оставаться стабильным длительное время, Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) прогрессирует в течение нескольких месяцев.
Характерным лабораторным изменением при Т-ПЛЛ является высокий лейкоцитоз, который может достигать 1000 • 10 9 /л.
По данным обследования более 100 пациентов Е. Matures и соавт. представили основные клинико-лабораторные проявления Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ): спленомегалия — 73 %; лимфаденопатия — 53 %; гепатомегалия — 40%; поражение кожи — 27 %; лейкоцитоз более 100 • 109/л — 75 %; анемия и тромбоцитопения — 30 %.
Этиологические причины Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ) не установлены. В сыворотке крови больных не обнаружены антитела к вирусам HTLV-I/II даже у пациентов из эндемичных регионов. С помощью анализа ДНК не удалось доказать наличие геномной последовательности вируса HTLV-I в опухолевых клетках.
Морфологическим субстратом Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ) более чем в 2/3 случаев являются пролимфоциты. Они имеют несколько больший по сравнению с обычным лимфоцитом размер, ядро с конденсированным хроматином, которое часто имеет неровные очертания и нуклеолу. Цитоплазма опухолевых клеток базофильная и не содержит гранул. У 20 % больных Т-ПЛЛ опухолевые клетки меньшего размера, в большинстве из них при световой микроскопии нуклеола видна плохо, однако электронно-микроскопическое исследование позволяет выявить ультраструктурные особенности, присущие пролимфоцитам. Такие случаи относят к мелкоклеточному варианту Т-ПЛЛ.
При иммунологическом фенотипировании пролимфоциты, как правило, имеют иммунофенотип CD2+CD5+ и выраженно экспрессируют антиген CD7. Количество CD7-антигенных детерминант на поверхности опухолевых клеток при Т-клеточном пролимфоцитарном лейкозе (Т-ПЛЛ) значительно больше, чем на нормальных Т-лимфоцитах и лимфоцитах при других посттимических Т-клеточных лимфопролиферативных заболеваниях. В 20 % случаев на мембране пролимфоцитов не экспрессируется CD3, однако этот маркер всегда обнаруживается в цитоплазме клеток. Применительно к экспрессии CD4 и CD8 уникального фенотипа, присущего исключительно Т-ПЛЛ, не существует.
Цитогенетические исследования, проведенные при Т-клеточном пролимфоцитарном лейкозе (Т-ПЛЛ), позволили выявить аномалии хромосом 14, 8 и 11. Перестройки хромосомы 14 составляют 2/3 всех цитогенетических изменений: инверсия хромосомы 14 — invl4(q11q32), тандемная транслокация между двумя хромосомами t(14; 14). Инверсия хромосомы 14 крайне редко встречается при других зрелоклеточных лимфопролиферативных заболеваниях Т-клеточной природы и считается патогномоничной для Т-клеточного пролимфоцитарного лейкоза. Важно отметить сходство цитогенетических изменений в опухолевых клетках при Т-ПЛЛ и в Т-лимфоцитах больных атаксией-телеангиэктазией. Описанные случаи развития Т-клеточных лейкозов у этих пациентов относятся к Т-ПЛЛ.
Как отмечалось, Т-клеточный пролимфоцитарный лейкоз является заболеванием с агрессивным течением. Больные Т-ПЛЛ обычно резистентны к стандартным схемам лечения, включающим алкилирующие препараты (хлорамбуцил, циклофосфамид). Включение в схему терапии антрациклинов (CHOP) позволяет получить ответ, чаще всего частичный и непродолжительный, только у 1/3 больных. Одним из наиболее активных цитостатических препаратов в лечении Т-клеточного пролимфоцитарного лейкоза является 2-деоксикоформицин (пентостатин). Использование его в дозе 4 мг/м2 еженедельно до достижения максимального эффекта позволяет получить общий ответ в 40 % случаев и только в 12 % случаев достигается полная ремиссия. В последние годы предпринимаются успешные попытки использовать при Т-ПЛЛ анти-CD52 моноклональное антитело (Campath-1H).
Иммунотерапия Campath-1H позволяет получить полную ремиссию более чем у половины пациентов, включая резистентных к деоксикоформицину.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Т-лимфотропный вирус человека (англ. Human T-lymphotropic virus, HTLV) - серотип вида Т-лимфотропного вируса из рода дельта-ретровирусов (Deltaretrovirus). Связан с такими злокачественными новообразованиями лимфоидной и кроветворной тканей, как Т-клеточный лейкоз и Т-клеточная лимфома. Более короткое название – HTLV 1 и 2 типа. Обследование на антитела к HTLV позволяет выявить инфицированных людей. Инфицирование бывает бессимптомным и может протекать в таком виде долгое время, человек при этом является потенциальным распространителем инфекции (при переливании крови или донорстве органов).
Синонимы русские
Антитела при Т-клеточной лимфоме, антитела при HTLV I - ассоциированной миелопатии (тропическом спастическом парапарезе), вирус HTLV I и II типа (ретровирусы типа С).
Синонимы английские
Метод исследования
Иммуноферментный анализ (ИФА).
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Т-лимфотропный вирус человека (англ. Human T-lymphotropic virus, HTLV) - серотип вида Т-лимфотропного вируса из рода дельта-ретровирусов (Deltaretrovirus). Связан с такими злокачественными новообразованиями лимфоидной и кроветворной тканей, как Т-клеточный лейкоз и Т-клеточная лимфома.
Т-лимфотропный вирус представляет собой штамм вируса, поражающий в основном взрослых людей. Вероятно, что он принимает участие в патогенезе некоторых демиелинизирующих заболеваний, например тропического спастического парапареза. Геном Т-лимфотропного вируса человека является диплоидным и состоит из двух копий одноцепочечных РНК, на которых в организме хозяина синтезируется одноцепочечная и далее двуцепочечная ДНК. Двуцепочечная ДНК далее интегрируется в геном хозяина в виде провируса.
HTLV I - Т-лимфотропный вирус человека первого типа (HTLV-I), также известный как вирус Т-клеточной лимфомы взрослых (ТЛВЧ-1), ассоциирован с такими заболеваниями, как HTLV-I-связанная миелопатия, гиперинфекция, вызванная круглым червём Strongyloides stercoralis, а также вирусная лейкемия. По некоторым данным, у 4-5 % заражённых появляются злокачественные опухоли в результате активности этих вирусов.
HTLV-II - Т-лимфотропный вирус человека второго типа (ТЛВЧ-2, HTLV-II) близкородственен Т-лимфотропному вирусу человека первого типа, имеет гомологию генома около 70 % по сравнению с HTLV-I.
Было выяснено, что возбудителем острого Т-клеточного лейкоза у человека являлся вирус, который назвали вирусом Т-клеточной лейкемии человека - HTLV I. По существующей классификации он был отнесен к классу ретровирусов.
HTLV-1 стал первым обнаруженным ретровирусом человека и был отнесен к подклассу онковирусов, т.е. вирусов, вызывающих рак. Несмотря на то что большинство попыток выделить ретровирусы из опухолевых клеток человека оказались безуспешными, установлено, что, по крайней мере, один вид ретровирусов вызывает злокачественное новообразование у человека. Это Т-лимфотропный вирус человека типа 1 - возбудитель Т-клеточного лейкоза - лимфомы взрослых. В отличие от онкогенных ретровирусов животных, Т-лимфотропный вирус человека типа 1 не содержит онкогенов, а его трансформирующие свойства связывают с белком Tax.
Т-лимфотропный вирус человека типа 1 передается от матери к ребенку (особенно через молоко), при половых контактах (чаще от мужчины к женщине), а также при переливании инфицированной крови и использовании инфицированных игл. Чаще всего заражение происходит в перинатальном периоде. В отличие от ВИЧ, который может передаваться с бесклеточным материалом, Т-лимфотропный вирус человека типа 1 менее заразен и для его передачи обычно необходим контакт между клетками.
Т-лимфотропный вирус человека типа 1 широко распространен в юго-западной части Японии и на острове Окинава, где заражено более 1 млн человек. Несмотря на высокий риск заражения, здесь выявляют только 500 случаев Т-клеточного лейкоза-лимфомы взрослых ежегодно.
Хотя ранние эпидемиологические исследования выявили растущее число носителей антител к Т-лимфотропному вирусу человека типа 1 среди инъекционных наркоманов, применение более специфичных методов серодиагностики показало, что в подавляющем большинстве случаев инфекция у инъекционных наркоманов обусловлена Т-лимфотропным вирусом человека типа 2.
Т-клеточный лейкоз-лимфома взрослых редко возникает у лиц, инфицированных при переливании компонентов крови; в то же время около 20 % больных тропическим спастическим парапарезом заражается через кровь. Развитие прогрессирующей спастической или атаксической миелопатии у носителей антител к Т-лимфотропному вирусу человека типа 1, вероятно, обусловлено прямым воздействием вируса на нервную систему; похожее заболевание может быть вызвано ВИЧ или Т-лимфотропным вирусом человека типа 2. Изредка у больных с тропическим спастическим парапарезом антитела к вирусу отсутствуют в сыворотке, но обнаруживаются в спинномозговой жидкости.
У носителей Т-лимфотропного вируса человека типа 1 вероятность заболеть в течение жизни Т-клеточным лейкозом-лимфомой взрослых составляет 2-5%, такой же риск развития тропического спастического парапареза. Эти заболевания встречаются только там, где распространен Т-лимфотропный вирус человека типа 1, причем у 95 % больных в сыворотке присутствуют антитела к этому вирусу.
Т-клеточный лейкоз-лимфома взрослых развивается через много (20 и более) лет после заражения. В половине случаев тропического спастического парапареза продолжительность латентного периода чаще составляет около 3 лет.
T-лимфотропный вирус человека типа 2, возможно, вызывает небольшую часть случаев волосатоклеточного лейкоза и различных Т-клеточных лимфом и лейкозов. Хотя Т-лимфотропный вирус человека типа 2 был выделен у одного больного с Т-клеточным вариантом волосатоклеточного лейкоза, его этиологическую связь с каким-либо заболеванием доказать не удалось. Однако, по некоторым данным, Т-лимфотропный вирус человека типа 2 может играть роль в развитии ряда заболеваний нервной системы, крови и кожи. Эти факты требуют проверки, особенно с учетом нечеткого различения Т-лимфотропных вирусов человека типа 1 и 2 в ранних исследованиях.
Инфекция HTLV-II эндемична для коренного населения Южной Америки, кроме того, встречается повсеместно у лиц, употребляющих наркотики внутривенно.
Вирус HTLV-II впервые был выделен от больного с волосатоклеточной лейкемией, однако с тех пор способность вируса вызывать лимфопролиферативные заболевания не подтвердилась. Патогенез HTLV-II-инфекции связывают с развитием HAM/TSP и других неврологических синдромов, а также пневмонии, бронхита и артрита. В Европе и США HTLV-II часто выявляют у ВИЧ-инфицированных лиц.
Лабораторная диагностика HTLV-I/II-инфекции основана на выявлении антител к вирусам, серологические свойства которых имеют значительное сходство; для скрининга используются методы ИФА и агглютинации латексных частиц. Подтверждающий иммуноблотинг с применением рекомбинантных антигенов позволяет различить эти две инфекции. Для уточнения диагноза дополнительно используется метод ПЦР; количественный вариант ПЦР позволяет оценить вирусную нагрузку, которая коррелирует с вероятностью развития ATL и TSP у носителей HTLV-I.
Сходство в строении Т-лимфотропных вирусов человека типа 1 и 2 до недавнего времени препятствовало созданию высокоспецифичных серологических методов, поэтому при эпидемиологических исследованиях нередко эти вирусы не разделяли. В результате сложилось ошибочное мнение, что среди инъекционных наркоманов преобладают носители Т-лимфотропного вируса человека типа 1. Однако обследование больших групп инъекционных наркоманов с применением высокоспецифичных серологических методов показало, что в подавляющем большинстве случаев они заражены Т-лимфотропным вирусом человека типа 2. Поскольку среди инфицированных Т-лимфотропным вирусом человека типа 2 значительно преобладают женщины, сделан вывод, что этот вирус легче передается от мужчин к женщинам, чем от женщин к мужчинам.
Обнаружение заболевания основывается на серологической диагностике инфицирования вирусом HTLV I и II типа и состоит в скрининговом обследовании, за которым следуют подтверждающие и уточняющие тесты. Обычно в качестве скрининговых тестов применяют иммуноанализ (ИФА), при получении повторно реактивных результатов в скрининговых тестах проводят подтверждение. Подтверждающими тестами для результатов ИФА тестов могут быть вестерн-блот или радиоиммунопреципитация.
Инфекционные причины лейкозов - лимфома Беркитта, иммунобластная лимфома, Т-клеточный лейкоз взрослых (ТКЛВ)
В экспериментальной лейкозологии существует много видов вирусов, с помощью которых индуцируют различные формы лейкозов. Известны также вирусы, вызывающие лейкозы у разных видов животных в естественных условиях. Для этих вирусов доказаны вертикальная трансмиссия (передача от матери потомству) и значительное распространение носительства в популяциях животных без признаков заболевания.
Эндемические формы инфекционных гемобластозов
Для большинства эндемических форм гемобластозов человека с определенностью установлено участие в индукции заболевания инфекционных агентов. В отношении некоторых убиквитарных форм существуют лишь более или менее убедительные подозрения. Рабочая группа МАИР в настоящее время причисляет к доказанным этиологическим факторам (группа 1) лимфопролиферативных гемобластозов следующие инфекционные агенты: вирус Эпштейна—Барр (EBV), вирус Т-клеточной лимфомы/лейкоза взрослых (HTLV-1), вирус иммунодефицита человека ВИЧ (HIV) и бактерию Helicobacter pylori.
Носительство EBV распространено очень широко и охватывает до 90 % взрослого населения. Вирус крайне неустойчив во внешней среде, поэтому заражение возможно только при прямом контакте — воздушно-капельным путем через слюну. Возраст, в котором происходит инфицирование, зависит от социальных условий: в развитых странах до трехлетнего возраста инфицируется около 20 % детей, тогда как в развивающихся —до 70 %. EBV обладает высокой тропностью к эпителию ротоглотки, где в нормальных условиях происходит его репродукция, и к В-лимфоцитам, в которых осуществляется бессимптомное носительство в виде кольцевой ДНК (эписома).
Доля содержащих эписому В-лимфоцитов невелика и довольно постоянна — 1 клетка на 10 5 —10 6 . В продуктивной фазе вирусная ДНК в В-клетках приобретает линейную форму.
В экваториальной зоне Африки с EBV связана лимфома Беркитта (ЛБ), которая занимает первое место по распространенности среди гемобластозов у детей на территориях, расположенных до 1550 м над уровнем моря в теплом и влажном климате. Особенно часто заболевание развивается у детей 5—13 лет; в 95 % всех случаев болезнь возникает до 16 лет. У детей моложе 2 лет заболевание практически не встречается. Классическое начало ЛБ: быстро растущая опухоль челюсти или органов брюшной полости у ребенка 5—8 лет. Мальчики преобладают в соотношении (1,7—2,0):1. Неберкиттовские лимфомы в этом регионе исключительно редки.
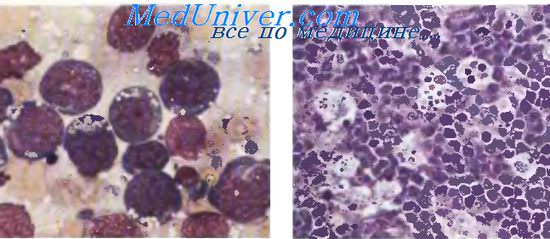
Морфология лимфомы Беркитта
Повышенное внимание к признакам лимфомы Беркитта (ЛБ) привело к тому, что это новообразование стали обнаруживать в западных странах среди НХЛ детского возраста (до 20 %). Раболеваемость в Африке значительно выше — в среднем 8 на 100 тыс. детей против 0,2 среди белого населения США.
Практически у 100 % больных лимфомой Беркитта (ЛБ) в африканских очагах в клетках опухоли выявляются включения ДНК EBV и определяются высокие титры антител к капсидным и ранним антигенам. Для здоровых детей с антителопродукцией к EBV риск ЛБ повышен в 50—60 раз. Среди спорадических случаев ЛБ носительство ДНК EBV в клетках опухоли много реже — около 30 %. В отличие от общего пула В-клеток ДНК ЕВУ содержится во всех опухолевых клетках ЛБ, причем в моноклоновой форме, что является основным подтверждением этиологической роли вируса. Известна способность EBV к трансформации клетки и индукции бесконечного деления В-лимфоцитов in vitro, тем не менее его роль в патогенезе ЛБ не вполне ясна.
Есть данные, что кофактором в развитии заболевания служит малярийный плазмодий: ареал высокого риска ЛБ совпадает, как правило, с эндемическими регионами малярии. У переселившихся в эти регионы из благополучных районов повышается вероятность возникновения обоих заболеваний. Проведение тотальной лекарственной профилактики малярии приводило к достоверному снижению заболеваемости ЛБ, а после прекращения противомалярийных мероприятий заболеваемость возвращалась на прежний уровень. Эпидемиологические наблюдения позволили построить следующую схему патогенеза ЛБ: малярийная инвазия, хроническая инфекция и, возможно, другие факторы могут служить активаторами пролиферации лимфоцитов.
Контакт с EBV вводит В-лимфоцит в бесконечный цикл деления (нечто близкое к инфекционному мононуклеозу). На следующем этапе случайная хромосомная транслокация способна вывести одну из активированных клеток из-под контроля, и она становится родоначальницей клона.
Анализ 665 случаев лимфомы Беркитта (ЛБ) из регистра Ибадана (Нигерия) показал значимое снижение относительной частоты заболеваний в период 1973—1990 гг. (37,1 % от всех опухолей у детей) по сравнению с 1960—1972 гг. (51,5 %). По всей видимости, это отражает реальное снижение заболеваемости за счет улучшения условий жизни и успешного контроля малярии.
Т-клеточный лейкоз взрослых (ТКЛВ) — заболевание, характерное для юго-восточных областей Японии (Окинава, Киуши, Шикоку), Экваториальной Африки и Карибских островов, Южной Америки и Ближнего Востока. В эндемических очагах до 20 % населения продуцируют антитела к HTLV-1 (среди населения США — 0,025 %). Наиболее распространен HTLV-1 в Африке —до 10 млн инфицированных, всего в мире их 15 — 20 млн. Трансмиссия вируса происходит вертикально (при грудном вскармливании ребенка), а также при гемотрансфузиях. Выявляемость носителей увеличивается с возрастом и достигает пика к 50 годам, причем доля женщин больше. Один случай ТКЛВ в год возникает среди приблизительно 1500 носителей HTLV-1.
В итоге заболевание развивается у 1—4 % носителей через 20— 30 лет после заражения, причем более 90 % заболевших серопозитивны к HTLV-1. Максимальное число заболевших регистрируется в Японии в возрасте около 60 лет, а в Африке — в 40—45 лет. Случаи заболевания детей исключительно редки, частота заболеваний у мужчин и женщин примерно равная. Смертность от ТКЛВ среди инфицированных достигает у мужчин 68, а у женщин 36 на 100 тыс.; среди серонегативных лиц она много ниже. Обследованные в эндемических районах матери заболевших ТКЛВ оказались инфицированными HTLV-1 в 100 %, тогда как матери больных другими формами лимфом — в 30 % случаев.
В Африке выявлено широкое распространение родственного вируса HTLV-2, который подозревается в инициации В-клеточных лейкозов.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Т-клеточный лейкоз-лимфома взрослых - опухоль из лимфоцитов CD4, вызванная Т-лимфотропным вирусом человека типа 1 (HTLV-I). Характерны поражение кожи и внутренних органов, резорбция костной ткани и гиперкальциемия. В крови обнаруживают атипичные лимфоциты.
Заболевания регистрируются главным образом на юге Японии, реже на островах Карибского бассейна, побережье Тихого океана, в Южной Америке, Экваториальной Африке и на севере США. В основном болеют взрослые негры и японцы. Мужчины заболевают чаще, чем женщины. Антитела к возбудителю часто находят в крови у наркоманов.
Что провоцирует / Причины Т-клеточного лейкоза-лимфомы взрослых:
Т-лимфотропный вирус человека типа 1 относится к семейству ретровирусов. Опухолевые клетки представляют собой активированные лимфоциты CD4, в избытке экспрессирующие а-цепи рецептора интерлей-кина-2. Опухоль развивается примерно у 5% инфицированных, у остальных наблюдается носительство провируса в лимфоцитах CD4. Поэтому полагают, что в патогенезе Т-клеточного лейкоза-лимфомы взрослых участвуют еще какие-то факторы. После заражения часть лимфоцитов CD4 приобретает способность к неограниченному размножению; отмечаются также повышенная митотическая активность, накопление генетических дефектов и дефицит клеточного иммунитета. Основная роль в развитии этих нарушений отводится вирусному белку tax.
Предполагается генетически обусловленная предрасположенность к заболеванию, однако нельзя исключить и возможность провоцирующего влияния каких-то факторов окружающей среды.
Симптомы Т-клеточного лейкоза-лимфомы взрослых:
Опухоль проявляется генерализованным увеличением лимфоузлов, гепатоспленомегалией, поражением кожи, остеолизом. Характерны гиперкальциемия, повышение активности ЛДГ в сыворотке. Опухолевые клетки полиморфны, экспрессируют CD4. Поражение кожи бывает представлено папулами, бляшками, опухолевидными образованиями, изъязвлениями. Инфильтрация костного мозга незначительна, анемия и тромбоцитопения нехарактерны.
Опухоль неуклонно прогрессирует, лечение малоэффективно.
Полихимиотерапия позволяет 50-70% больных достичь полной ремиссии, однако у половины из них ремиссия длится менее 12 мес.
Из-за глубокого иммунодефицита очень высока частота вторичных инфекций, многие из которых обусловлены условно-патогенными микроорганизмами.
Описана также хроническая форма заболевания - с поражением кожи, но без гепатоспленомегалии и увеличения лимфоузлов. Характерен умеренный лимфоцитоз, доля опухолевых клеток в крови невелика. Продолжительность жизни таких больных может достигать нескольких лет - пока болезнь не перейдет в острую форму.
Выделяют четыре формы Т-клеточного лейкоза-лимфомы взрослых: острую, лимфоматозную, хроническую и тлеющую. При любой форме заболевания опухоль развивается за счет моноклональной пролиферации лимфоцитов CD4. Во всех таких клетках провирус встроен в ДНК одинаково и обнаруживается уникальная перестройка генов, кодирующих антигенраспознающие рецепторы Т-лимфоцитов.
Острая форма встречается в 60% случаев; заболевание характеризуется коротким продромальным периодом (от появления первых симптомов до постановки диагноза проходит около 2 нед) и бурным течением (продолжительность жизни - 6 мес). Клинические проявления: быстро прогрессирующие поражение кожи и поражение легких, гиперкальциемия и лимфоцитоз. Появляются атипичные лимфоциты с дольчатыми ядрами или атипичные лимфоциты с ядрами в виде раздвоенного копыта . В ДНК опухолевых клеток встроен провирус, а на их поверхности экспрессируются рецепторы CD4, CD3 и CD25 (низкоаффинные рецепторы ИЛ-2). Уровень CD25 в сыворотке служит опухолевым маркером. Анемия и тромбоцитопения наблюдаются редко. Поражения кожи иногда трудно отличить от сыпи при грибовидном микозе. Часто возникающие очаги лизиса костной ткани обычно содержат не опухолевые клетки, а остеокласты. Остеогенез в таких очагах подавлен. Поражение костного мозга в большинстве случаев носит очаговый характер, хотя при цитологическом исследовании обнаруживают бластные клетки.
Гиперкальциемия при Т-клеточном лейкозе-лимфоме взрослых вызвана несколькими причинами. Опухолевые клетки продуцируют факторы активации остеокластов (ФНОальфа, ФНОбета, ИЛ-1), а также способны вырабатывать ПТГ-подобные пептиды. Заболевание сопровождается иммунодефицитом , на фоне которого возникают оппортунистические инфекции, аналогичные тем, что встречаются при СПИДе. Патогенез иммунодефицита не установлен. Изменения на рентгенограмме грудной клетки в половине случаев обусловлены лейкозной инфильтрацией легких, а остальное приходится на пневмонии, вызванные условно-патогенными возбудителями (Pneumocystis carinii и другими грибами). Желудочно-кишечные нарушения практически всегда связаны с оппортунистической инфекцией. В сыворотке нередко повышены активности ЛДГ и ЩФ. Примерно у 10% больных наблюдаются симптомы лептоменингита: слабость, психические нарушения, парестезия и головная боль. В отличие от других лимфом, поражающих ЦНС, при Т-клеточном лейкозе-лимфоме взрослых содержание белка в СМЖ может оставаться в норме. Диагноз подтверждает присутствие в СМЖ опухолевых клеток.
Лимфоматозная форма развивается у 20% больных. По клинической картине и течению данная форма напоминает острую, но отличается малым количеством атипичных лимфоцитов в крови и выраженным увеличением лимфоузлов. Гистологическая картина разнообразна: опухоли свойственен выраженный клеточный и ядерный полиморфизм. Однако течение болезни не зависит от гистологического строения опухоли. Рождение больного в эндемическом районе, характерное поражение кожи и гиперкальциемия - признаки, позволяющие поставить предварительный диагноз, который подтверждается при обнаружении в сыворотке антител к Т- лимфотропному вирусу человека типа 1.
При хронической форме ЦНС, кости и ЖКТ обычно не поражаются, а концентрация кальция и активность ЛДГ в сыворотке остаются нормальными. Обычно продолжительность жизни больных - 2 года. Иногда хроническая форма переходит в острую.
Тлеющая форма встречается не более чем у 5% больных. ДНК моноклональных опухолевых клеток содержит встроенный провирус; доля атипичных лимфоцитов в крови - менее 5%; гиперкальциемия, лимфаденопатия и гепатоспленомегалия, а также изменения со стороны ЦНС, костей и ЖКТ отсутствуют, но легкие и кожа могут поражаться. Обычно продолжительность жизни больных - 5 лет и более.
Течение и прогноз
При хронической и тлеющей формах Т-клеточного лейкоза-лимфомы взрослых единственными симптомами болезни могут быть инфильтрация кожи и небольшой лимфоцитоз в крови и костном мозге. Острая и лимфоматозная формы характеризуются бурным течением, тяжелым поражением кожи, легких и костей. При нормальном уровне кальция в крови средняя продолжительность жизни составляет 50 нед с момента постановки диагноза, а при гиперкальциемии - 12,5 нед (от 2 нед до 1 года). Причины смерти: оппортунистические инфекции, ДВС-синдром.
Диагностика Т-клеточного лейкоза-лимфомы взрослых:
Клиническая картина и обнаружение антител к Т-лимфотропному вирусу человека типа 1. Диагноз подтверждают с помощью молекулярно-генетического исследования (в ДНК пораженных лимфоцитов CD4 встроена ДНК провируса).
Дополнительные исследования
Общий анализ крови
Количество лейкоцитов от нормального до 500 000. В мазке крови - атипичные лимфоциты с дольчатыми ядрами, похожие на клетки Сезари.
Патоморфология кожи
В верхних и средних слоях дермы выявляют периваскулярные или диффузные инфильтраты из крупных атипичных лимфоцитов; эпидермис обычно не затронут. Иногда инфильтраты в дерме плотные, а в эпидермисе встречаются микроабсцессы Потрие, состоящие из большого количества крупных атипичных лимфоцитов, среди которых попадаются гигантские клетки.
Биохимический анализ крови Гиперкальциемия: в начале заболевания - у 25% больных, в дальнейшем - более чем у половины.
Серологические реакции Антитела к Т-лимфотропному вирусу человека типа 1 выявляют с помощью иммуно-ферментного анализа и иммуноблотткнга. Среди инъекционных наркоманов, зараженных ВИЧ, около 30% одновременно инфицированы Т-лимфотропным вирусом человека типа 1.
Лечение Т-клеточного лейкоза-лимфомы взрослых:
Используют различные комбинации противоопухолевых средств. Ремиссии непродолжительные, достигаются менее чем в 30% случаев. Острая и лимфоматозная формы болезни к стандартным схемам химиотерапии не чувствительны. Недавно получены обнадеживающие результаты при комбинированном лечении зидовудином (внутрь) и интерфероном а (п/к).
Профилактика Т-клеточного лейкоза-лимфомы взрослых:
Для предотвращения дальнейшего распространения инфекции обследуют всех членов семьи и половых партнеров больного. Серопозитивные носители не должны становиться донорами.
К каким докторам следует обращаться если у Вас Т-клеточный лейкоз-лимфома взрослых:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Т-клеточного лейкоза-лимфомы взрослых, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Хронические Т-клеточные лейкозы - Т-клеточная лейкемия, лимфома кожи, синдром Сезари
Т-клеточный крупногранулярный лимфоцитарный лейкоз встречается в 30-50 раз реже В-клеточного ХЛЛ и развивается в возрасте 50-55 лет, чаще у женщин. Основной морфологический признак заболевания — наличие больших гранулярных (содержащих азурофильные гранулы) лимфоцитов в периферической крови и костном мозге. Критерий диагностики — обнаружение больше 2 • 109/л больших гранулярных лимфоцитов в периферической крови. Наиболее частый иммунофенотип: CD3+, CD8+, CD4-, TCRab+.
Лейкемические клетки экспрессируют маркеры апоптоза (Fas или CD95 и Fas-лиганд), но резистентны к Fas-индуцируемому апоптозу.
Спленомегалия выявляется у 20% пациентов, лимфаденопатия и гепа-томегалия встречаются еще реже. В связи с выраженной нейтропенией нередко возникают рецидивирующие инфекции. У 30% больных наблюдается ассоциация с аутоиммунными заболеваниями (аутоиммунная гемолитическая анемия, ревматоидный артрит и др.).
Течение заболевания вариабельно, стандартное лечение не разработано.
Т-клеточная лейкемия/лимфома взрослых
Т-клеточная лейкемия/лимфома взрослых — редкое лимфопролиферативное заболевание, встречающееся преимущественно в странах бассейна Карибского моря и Японии. Доказанным этиологическим фактором является ретровирус HTLV-1. В эндемичных районах инфицировано 5% населения; в течение жизни заболевает один человек из 50-100 инфицированных (в Японии при наличии около 1 миллиона вирусоносителей в год регистрируется около 500 случаев заболевания). Спорадические случаи Т-клеточной лейкемии/лимфомы взрослых отмечены в Европе и Северной Америке.
Опухолевые клетки полиморфны, с полисегментированными, похожими на лепестки цветка, ядрами. Иммунофенотип клеток: CD7-, CD2+, CD3+, CD4+, CD5+, CD25+. Наиболее частые цитогенетические находки — трисомия 12, del 6q.
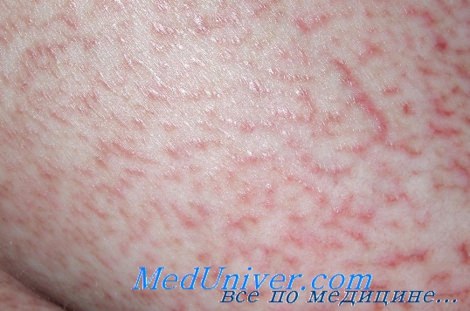
Грибовидный микоз
Т-клеточные лимфомы кожи - грибовидный микоз, синдром Сезари
Т-клеточные лимфомы кожи — гетерогенная группа лимфопролиферативных заболеваний с первичным поражением кожи. В последние десятилетия заболеваемость Т-клеточными лимфомами кожи достоверно увеличилась. В большинстве случаев встречаются грибовидный микоз и синдром Сезари. Реже наблюдаются первичные СD30-позитивные Т-клеточные лимфопролиферативные заболевания: первичная кожная анапластическая крупноклеточная лимфома (КМ+) и лимфоматоидный папулез.
Грибовидный микоз многие годы протекает с изолированным поражением кожи (эритема, папулы, бляшки, эритродермия), однако в дальнейшем в большинстве случаев развивается агрессивная Т-клеточная лимфома с поражением лимфатических узлов, селезенки, печени и других органов и неблагоприятным исходом. При этом в крови и костном мозге часто обнаруживаются клетки Сезари (перекрестный синдром).
Синдром Сезари — генерализованная Т-клеточная лимфома, характеризующаяся поражением кожи, лимфоаденопатией и наличием в периферической крови неопластически трансформированных Т-лимфоцитов — клеток Сезари (атипичные лимфоциты с большим ядром неправильной формы и скудной базофильной цитоплазмой).
Синдром Сезари характеризуется сходным с грибовидным микозом поражением кожи, однако заболевание сопровождается ранней генерализацией и неблагоприятным течением (лишь 20% больных живут более 5 лет).
Диагноз лимфом кожи доказывается при морфологическом исследовании и результатами иммунофенотипирования (особенно важно это при CD30-позитивных лимфомах). При грибовидном микозе и синдроме Сезари нередко выявляются различные нарушения кариотипа (прежде всего при прогрессировании заболевания). При молекулярно-генетическом исследовании у большинства больных Т-клеточными лимфомами кожи обнаруживается клональная реаранжировка генов Т-клеточных рецепторов.
При локализованных формах Т-клеточных лимфом кожи применяется местное лечение: PUVA-терапия (ультрафиолетовое облучение с фотосенсибилизатором), локальное облучение и введение цитостатиков в опухолевые очаги. При синдроме Сезари используются монохимиотерапия и полихимиотерапия, однако частота ремиссий невелика и они обычно непродолжительны.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: