
Болезнь кори гликогеноз студопедия
Обновлено: 13.05.2024
Гликогеноз типа I - заболевание, описанное Гирке в 1929 г., однако ферментный дефект был установлен Кори только в 1952 г. Гликогеноз типа I встречается у 1 из 200 000 новорожденных. Заболеваемость мальчиков и девочек одинакова. Наследование аутосомно-рецессивное. При гликогенозе I типа ( болезнь Гирке) клетки печени и извитых почечных канальцев заполнены гликогеном, однако эти запасы оказываются недоступными: об этом свидетельствует гипогликемия, а также отсутствие повышения уровня глюкозы в крови в ответ на адреналин и глюкагон. Обычно у этих больных развиваются кетоз и гиперлипемия, что вообще характерно для состояния организма при недостатке углеводов. В печени, почках и тканях кишечника активность глюкозо-6-фосфатазы либо крайне низка, либо вообще отсутствует.
Патогенез (что происходит?) во время Гликогеноза I типа (болезни Гирке):
Заболевание обусловлено дефектами ферментной системы печени, превращающей глюкозо-6-фосфат в глюкозу. Нарушается как гликогенолиз, так и глюконеогенез, что приводит к гипогликемии голодания с лактацидозом, гиперурикемии и гипертриглицеридемии. В печени накапливается избыток гликогена.
Ферментная система, превращающая глюкозо-6-фосфат в глюкозу, содержит не менее 5 субъединиц: глюкозо-6-фосфатазу (катализирует гидролиз глюкозо-6-фосфата в просвете эндоплазматического ретикулума), регуляторный Са2(+)-связывающий белок и белки-переносчики (транслоказы), T1, T2 и T3, которые обеспечивают переход глюкозо-6-фосфата, фосфата и глюкозы через мембрану эндоплазматического ретикулума.
Дефект глюкозо-6-фосфатазы (гликогеноз типа Ia) и дефект глюкозо-6-фосфат-транслоказы (гликогеноз типа Ib) проявляются сходными клиническими и биохимическими нарушениями. Чтобы подтвердить диагноз и точно установить ферментный дефект, необходима биопсия печени и исследование активности глюкозо-6-фосфатазы.
Симптомы Гликогеноза I типа (болезни Гирке):
Клинические проявления гликогеноза типа I у новорожденных, грудных детей и детей старшего возраста неодинаковы. Причина - различия рациона и режима питания в этих возрастных группах.
Иногда в первые дни и недели жизни возникает гипогликемия голодания, однако в большей части случаев болезнь протекает бессимптомно, поскольку грудной ребенок часто питается и получает достаточное количество глюкозы. Нередко болезнь диагностируют через несколько месяцев после рождения, когда у ребенка обнаруживают увеличение живота и гепатомегалию. Бывают одышка и субфебрильная температура без признаков инфекции. Одышка вызвана гипогликемией и лактацидозом из-за недостаточной продукции глюкозы. Когда интервалы между кормлениями увеличиваются и ребенок начинает спать ночью, появляются симптомы гипогликемии, особенно по утрам. Тяжесть и длительность гипогликемии постепенно увеличиваются, что приводит к системным метаболическим нарушениям.
Если лечение не проводят, изменяется внешность ребенка. Характерны гипотрофия мышц и скелета, задержка роста и физического развития, отложение жира под кожей. Ребенок становится похож на больного с синдромом Кушинга. Развитие познавательных и социальных навыков не страдает, если только повторные приступы гипогликемии не вызвали повреждения головного мозга. Если ребенок не получает достаточного количества углеводов и гипогликемия голодания сохраняется, то задержка роста и физического развития становится резко выраженной. Некоторые дети с гликогенозом типа I умирают от легочной гипертензии.
Нарушение функции тромбоцитов проявляется повторными носовыми кровотечениями или кровоточивостью после стоматологических и других хирургических вмешательств. Отмечаются нарушения адгезии и агрегации тромбоцитов; нарушено также высвобождение АДФ из тромбоцитов в ответ на адреналин и контакт с коллагеном. Тромбоцитопатия вызвана системными метаболическими нарушениями; после лечения она исчезает.
УЗИ и экскреторная урография выявляют увеличение почек. У большинства больных выраженных нарушений функции почек не бывает, отмечается лишь повышение СКФ (скорость клубочковой фильтрации) . В очень тяжелых случаях может развиться тубулопатия с глюкозурией, фосфатурией, гипокалиемией и аминоацидурией (как при синдроме Фанкони). У подростков иногда наблюдается альбуминурия, а у молодых людей часто развивается тяжелое поражение почек с протеинурией, повышением АД (артериального давления) и падением клиренса креатинина, обусловленное фокально-сегментарным гломерулосклерозом и интерстициальным фиброзом. Эти нарушения приводят к терминальной почечной недостаточности.
Селезенка не увеличена.
Без лечения резко возрастают уровни свободных жирных кислот, триглицеридов и апопротеина C-III, который участвует в транспорте триглицеридови богатых триглицеридами липопротеидов. Уровни фосфолипидов и холестерина повышаются умеренно. Очень высокий уровень триглицеридов обусловлен их чрезмерной продукцией в печени и снижением их периферического метаболизма из-за снижения активности липопротеидлипазы. При тяжелой гиперлипопротеидемии на разгибательных поверхностях конечностей и ягодицах могут появляться эруптивные ксантомы.
Отсутствие лечения или неправильное лечение приводят к задержке роста и полового развития.
Аденомы печени по неизвестным причинам возникают у многих больных, обычно в возрасте 10-30 лет. Аденомы могут малигнизироваться, возможны кровоизлияния в аденому. На сцинтиграммах печени аденомы выглядят как участки пониженного накопления изотопа. Для обнаружения аденом применяют УЗИ. При подозрении на злокачественный рост более информативны МРТ (магнитно-резонансная томография) и КТ (компьютерная томография), позволяющие проследить превращение небольшого четко отграниченного новообразования в более крупное, с размытыми краями. Рекомендуется периодически измерять уровень альфа-фетопротеина в сыворотке (это маркер печеночноклеточного рака).
С возрастом тяжесть гипогликемии голодания уменьшается. Вес тела растет быстрее, чем вес головного мозга, поэтому соотношение между скоростью продукции и утилизации глюкозы становится более выгодным. Скорость продукции глюкозы возрастает за счет активности амило-1,6-глюкозидазы в печени и мышцах. В результате уровень глюкозы натощак постепенно повышается.
Клинические проявления гликогеноза типа Iа и типа Ib одинаковы, но при гликогенозе типа Ib наблюдается постоянная или преходящая нейтропения. В тяжелых случаях развивается агранулоцитоз. Нейтропения сопровождается дисфункцией нейтрофилов и моноцитов, поэтому повышается риск стафилококковых инфекций и кандидоза. У некоторых больных возникает воспалительное заболевание кишечника , напоминающее болезнь Крона.
Диагностика Гликогеноза I типа (болезни Гирке):
При лабораторной диагностике гликогеноза типа I проводятся:
- обязательные исследования: измеряют уровни глюкозы, лактата, мочевой кислоты и активность ферментов печени натощак; у новорожденных и грудных детей с гликогенозом типа I уровень глюкозы в крови после 3-4-часового голодания падает до 2,2 ммоль/л и ниже; если продолжительность голодания превышает 4 ч, уровень глюкозы почти всегда меньше 1,1 ммоль/л; гипогликемия сопровождается значительным повышением уровня лактата и метаболическим ацидозом; сыворотка обычно мутная или похожа на молоко из-за очень высокого содержания триглицеридов и умеренно повышенного содержания холестерина; отмечаются также гиперурикемия и повышение активности АсАТ (аспартатаминотрансферазы) и АлАТ (аланинаминотрансферазы).
- провокационные пробы: чтобы отличить гликогеноз типа I от других гликогенозов и точно определить ферментный дефект, у грудных детей и детей старшего возраста измеряют уровень метаболитов (глюкозы, свободных жирных кислот, кетоновых тел, лактата и мочевой кислоты) и гормонов (инсулина , глюкагона , адреналина, кортизола и СТГ (соматотропного гормона)) натощак и после приема глюкозы; схема исследования такова: ребенку дают глюкозу внутрь в дозе 1,75 г/кг, затем каждые 1-2 ч берут кровь; в каждой пробе быстро измеряют концентрацию глюкозы; последнюю пробу берут не позже чем через 6 ч после приема глюкозы либо в тот момент, когда концентрация глюкозы снизилась до 2,2 ммоль/л;
- провокационная проба с глюкагоном: глюкагон вводят в/м или в/в струйно в дозе 30 мкг/кг (но не более 1 мг) через 4-6 ч после еды или приема глюкозы; кровь для определения глюкозы и лактата берут за 1 мин до инъекции глюкагона и через 15, 30,45, 60,90 и 120 мин после инъекции. При гликогенозе типа I глюкагон не повышает либо незначительно повышает уровень глюкозы, тогда как исходно повышенный уровень лактата продолжает нарастать;
- специальное исследование: проводят биопсию печени, исследуют гликоген; содержание гликогена сильно увеличено, но структура его нормальная;
- специальные исследования для точного установления ферментного дефекта, лежащего в основе гликогеноза типа I: измеряют активность глюкозо-6-фосфатазы в цельных и разрушенных микросомах печени (по образованию глюкозы и фосфата из глюкозо-6-фосфата); микросомы разрушают повторным замораживанием и оттаиванием биоптата; при гликогенозе типа Iа активность глюкозо-6-фосфатазы не определяется ни в цельных, ни в разрушенных микросомах; при гликогенозе типа Ib активность глюкозо-6-фосфатазы в разрушенных микросомах нормальная, а в цельных микросомах отсутствует или сильно снижена (поскольку дефектная глюкозо-6-фосфат-транслоказа не переносит глюкозо-6-фосфат через мембраны микросом);
- методы молекулярной биологии (выявление генетического дефекта путем ПЦР (полимеразной цепной реакции) и последующей гибридизации со специфическими олигонуклеотидами).
Специальные исследования и методы молекулярной биологии доступны только специализированным лабораториям; в США, к примеру, в лабораториях: Dr. Y. Т. Chen, Division of Genetics and Metabolism, Duke University Medical Center, Durham, North Carolina, U.S.A.; Dr. R. Grier, Biocemical Genetics Laboratory, Nemours Children's Clinic, Jacksonville, Florida, U.S.A.
Лечение Гликогеноза I типа (болезни Гирке):
Метаболические нарушения при гликогенозе типа I, обусловленные недостаточной продукцией глюкозы , возникают уже через несколько часов после еды, а при длительном голодании значительно усиливаются. Поэтому лечение гликогеноза типа I сводится к частому кормлению ребенка. Цель лечения - предупредить падение концентрации глюкозы в крови ниже 4,2 ммоль/л - порогового уровня, при котором происходит стимуляция секреции контринсулярных гормонов .
Если ребенок своевременно получает достаточное количество глюкозы размеры печени уменьшаются, лабораторные показатели приближаются к норме, кровоточивость исчезает, рост и психомоторное развитие нормализуются.
К каким докторам следует обращаться если у Вас Гликогеноз I типа (болезнь Гирке):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Гликогеноза I типа (болезни Гирке), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Гликогенозы у детей: болезни Гирке, Форбса-Кори, Андерсена, Мак-Ардла, Таруи
Гликогеноз I типа (болезнь Гирке) — не истинная миопатия, а заболевание, обусловленное дефицитом печеночного фермента глюкозо-6-фосфатазы, который в норме не присутствует в мышце, тем не менее у детей с этим заболеванием выявляется гипотония и умеренно выраженная мышечная слабость неизвестной этиологии. Гликогеноз II типа (болезнь Помпе) — аутосомно-рецессивно унаследованный дефицит гликолитического лизосомного фермента кислой мальтазы. Из 12 известных типов гликогенозов только II тип обусловлен дефектом лизосомного фермента. Аномальный ген картирован в локусе 17q23. Описано две формы заболевания.
Младенческая форма характеризуется тяжелой генерализованной миопатией и кардиомиопатией. У пациентов выявляется кардиомегалия, гепатомегалия, диффузная гипотония и мышечная слабость. Активность КФК в крови значительно повышена. Мышечная биопсия выявляет вакуолярную миопатию в сочетании с нарушением активности ферментов лизосом, таких как кислая и щелочная фосфатазы. Смерть обычно наступает в младенческом или раннем детском возрасте.
Поздняя детская или взрослая форма представлена миопатией с более легким течением, без увеличения сердца и печени. Клинические проявления могут отсутствовать до позднего детского или раннего зрелого возраста, однако возможно появление признаков мышечной слабости (обусловленной миопатией) и гипотонии даже в раннем младенческом возрасте. Активность КФК в крови значительно повышена, результаты мышечной биопсии имеют диагностическое значение даже на пресимптомной стадии заболевания.
Диагноз гликогеноза II типа подтверждается при количественном анализе активности кислой мальтазы при биопсии мышц или печени. При редком варианте дефицита кислой мальтазы с легким течением ее активность при биопсии мышц может находиться на нижней границе нормы с периодическим снижением до субнормального уровня, при этом результаты мышечной биопсии напоминают гликогеноз II типа, но изменения выражены более умеренно.
Другая форма — болезнь Данона — характеризуется Х-сцепленным рецессивным типом наследования, аномальный ген картирован в локусе Xql4. В основе заболевания лежит первичный дефицит протеина-2 мембран лизосом (LAMP2), который приводит к развитию гипертрофической кардиомиопатии, миопатии с поражением мышц проксимальных отделов конечностей и умственной отсталости.
Гликогеноз III типа (болезнь Форбса-Кори) обусловлен дефицитом фермента, расщепляющего гликоген (амило-1,6-глюкозидаза). Это наиболее распространенный гликогеноз с наименее тяжелыми клиническими проявлениями по сравнению с другими типами гликогенозов. В младенческом возрасте часто встречаются такие симптомы, как гипотония, мышечная слабость, гепатомегалия, гипогликемия при исследовании крови натощак, однако эти симптомы часто спонтанно исчезают и в детском возрасте, а также у взрослых клинические проявления могут отсутствовать.
В других случаях отмечается медленное прогрессирование атрофии мышц дистальных отделов конечностей, цирроза печени и сердечной недостаточности. При мышечной биопсии обнаруживаются минимально выраженные миопатические изменения, включающие вакуолизацию мышечных волокон.
Гликогеноз IV типа (болезнь Андерсена) обусловлен дефицитом фермента, участвующего в синтезе гликогена, приводящего к синтезу аномальных молекул гликогена — амилопектина — в печени, ретикулоэндотелиальных клетках, скелетной мускулатуре и сердечной мышце. Гипотония, генерализованная мышечная слабость, атрофия мышц и контрактуры — характерные признаки миопатического процесса. Большинство пациентов умирает до 4-летнего возраста в связи с развитием печеночной или сердечной недостаточности. Описаны отдельные случаи заболевания у детей без признаков нервно-мышечного заболевания.
Гликогеноз V типа (болезнь Мак-Ардла) обусловлен дефицитом мышечной фосфорилазы, наследуемым по аутосомно-рецессивному типу, аномальный ген картирован в локусе 1lql3. Основным клиническим проявлением заболевания служит непереносимость физической нагрузки, которая вызывает болезненный мышечный спазм (крампи), мышечную слабость и миоглобинурию; однако между приступами мышечная сила не снижена. Активность КФК в крови повышена только во время физической нагрузки. Характерным клиническим признаком служит отсутствие наблюдаемого в норме повышения уровня лактата в крови во время физической нагрузки, приводящей к ишемии.
Это обусловлено невозможностью превращения пирувата в лактат при анаэробных состояниях in vivo. Дефицит миофосфорилазы можно обнаружить с помощью гистохимических и биохимических методов в мышечном биоптате.
Редкая неонаталъная форма дефицита миофосфорилазы вызывает бульбарные расстройства в раннем младенческом возрасте, которые могут быть настолько выражены, что приводят к летальному исходу в периоде новорожденное™. В других случаях возможно медленное прогрессирование мышечной слабости, напоминающее мышечную дистрофию.
Отдаленный прогноз благоприятный. Пациенты должны научиться контролировать свой уровень физической нагрузки; тяжелой инвалидизации вследствие хронической миопатии или поражения сердца не отмечается.
Гликогеноз VII (болезнь Таруи) представляет собой дефицит мышечной фосфофруктокиназы. Хотя это заболевание встречается реже, чем гликогеноз V типа, оба заболевания характеризуются непереносимостью физической нагрузки, похожим клиническим течением и невозможностью превращения пирувата в лактат. Биохимическое исследование мышечных биоптатов позволяет дифференцировать эти два типа гликогенозов. Заболевание наследуется по аутосомно-рецессивному типу, аномальный ген картирован в локусе lcenq32.
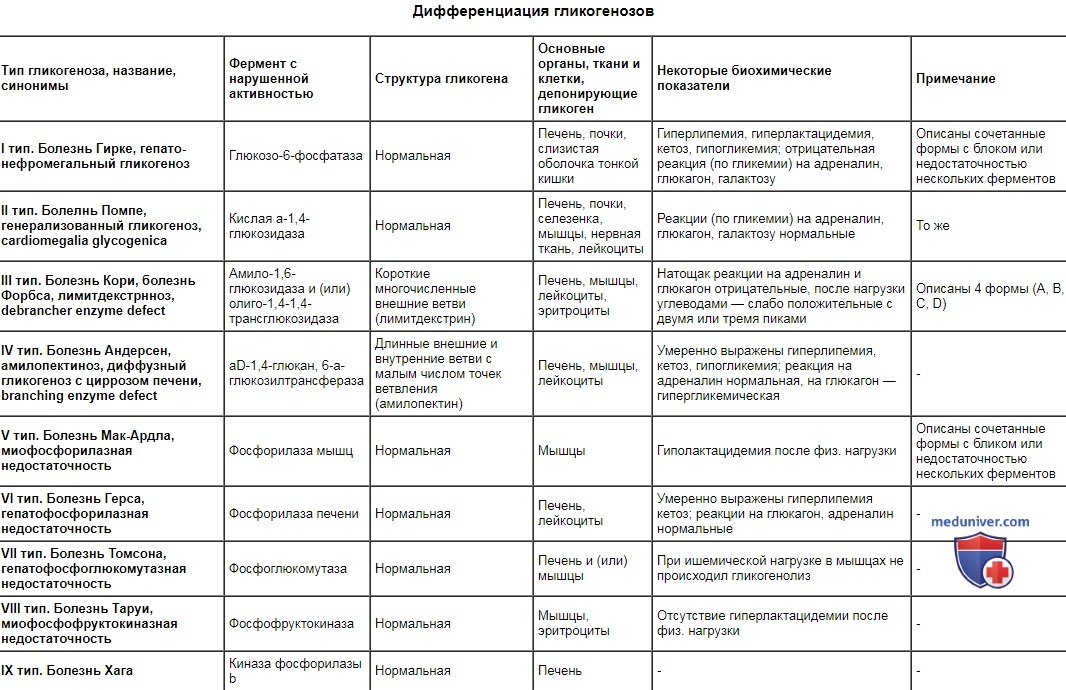
Дифференциация гликогенозов
| Тип гликогеноза, название, синонимы | Фермент с нарушенной активностью | Структура гликогена | Основные органы, ткани и клетки, депонирующие гликоген | Некоторые биохимические показатели | Примечание |
| I тип. Болезнь Гирке, гепато-нефромегальный гликогеноз | Глюкозо-6-фосфатаза | Нормальная | Печень, почки, слизистая оболочка тонкой кишки | Гиперлипемия, гиперлактацидемия, кетоз, гипогликемия; отрицательная реакция (по гликемии) на адреналин, глюкагон, галактозу | Описаны сочетанные формы с блоком или недостаточностью нескольких ферментов |
| II тип. Болелнь Помпе, генерализованный гликогеноз, cardiomegalia glycogenica | Кислая а-1,4-глюкозидаза | Нормальная | Печень, почки, селезенка, мышцы, нервная ткань, лейкоциты | Реакции (по гликемии) на адреналин, глюкагон, галактозу нормальные | То же |
| III тип. Болезнь Кори, болезнь Форбса, лимитдекстрнноз, debrancher enzyme defect | Амило-1,6-глюкозидаза и (или) олиго-1,4-1,4-трансглюкозидаза | Короткие многочисленные внешние ветви (лимитдекстрин) | Печень, мышцы, лейкоциты, эритроциты | Натощак реакции на адреналин и глюкагон отрицательные, после нагрузки углеводами — слабо положительные с двумя или тремя пиками | Описаны 4 формы (А, В, C, D) |
| IV тип. Болезнь Андерсен, амилопектиноз, диффузный гликогеноз с циррозом печени, branching enzyme defect | aD-1,4-глюкан, 6-а-глюкозилтрансфераза | Длинные внешние и внутренние ветви с малым числом точек ветвления (амилопектин) | Печень, мышцы, лейкоциты | Умеренно выражены гиперлипемия, кетоз, гипогликемия; реакция на адреналин нормальная, на глюкагон — гипергликемическая | - |
| V тип. Болезнь Мак-Ардла, миофосфорилазная недостаточность | Фосфорилаза мышц | Нормальная | Мышцы | Гиполактацидемия после физ. нагрузки | Описаны сочетанные формы с бликом или недостаточностью нескольких ферментов |
| VI тип. Болезнь Герса, гепатофосфорилазная недостаточность | Фосфорилаза печени | Нормальная | Печень, лейкоциты | Умеренно выражены гиперлипемия кетоз; реакции на глюкагон, адреналин нормальные | - |
| VII тип. Болезнь Томсона, гепатофосфоглюкомутазная недостаточность | Фосфоглюкомутаза | Нормальная | Печень и (или) мышцы | При ишемической нагрузке в мышцах не происходил гликогенолиз | - |
| VIII тип. Болезнь Таруи, миофосфофруктокиназная недостаточность | Фосфофруктокиназа | Нормальная | Мышцы, эритроциты | Отсутствие гиперлактацидемии после физ. нагрузки | - |
| IX тип. Болезнь Хага | Киназа фосфорилазы b | Нормальная | Печень | - | - |
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Гликогеноз Кори, Андерсена, Мак-Ардла. Болезнь Херса, Томсона, Таруи
Гликогеноз III типа, (болезнь Кори). При этом гликогенозе накапливается аномальный гликоген (лимитдекстрин) преимущественно в печени, а также в скелетной мускулатуре и в миокарде. Наследование аутосомно-рецессивное. Заболевание характеризуется отсутствием фермента амило-1,6-гликозидазы. В результате этого нарушается расщепление гликогена, образуются его молекулы с короткими внешними цепями. Клинические и патологоанатомические изменения сходны с гликогенозом I типа, но не так резко выражены. Дефект фермента амило-1,6-глюкозидазы обнаруживается в лейкоцитах крови, аномальный гликоген — в эритроцитах.
Гликогеноз IV типа, амилопектиноз (болезнь Андерсена). Очень редкий тип гликогеноза, при котором имеет место нарушение синтеза гликогена, описан Дороти Андерсен в 1956 г. Причина болезни — в отсутствии фермента амило-(1,4-1,6)-трансглюкозилазы (ветвящий фермент), который в норме участвует в образовании точек ветвления молекул гликогена. Заболевание проявляется к концу грудного возраста или в раннем детском возрасте. Ведущие симптомы: цирроз печени, гепато- и спленомегалия, асцит, желтуха, кровоточивость. Прогноз, как при любом циррозе, неблагоприятный.
Гликогеноз V типа (болезнь Мак-Ардла), Классический мышечный гликогеноз характеризуется дефектом мышечной фосфорилазы. Симптомы: мышечные болиг возникающие после мышечного напряжения. В покое и после приема глюкозы боли исчезают. Постепенно развивается тугоподвижность, мышечная слабость, моча темного цвета от присутствия миоглобина. Болезнь развивается в старшем детском возрасте (после 10 лет). В свежих крпостатных срезах реакция на фосфорилазу отрицательная.
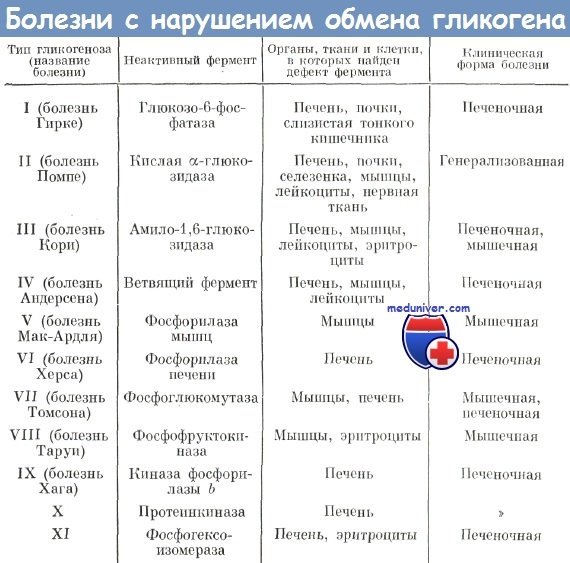
Гликогеноз VI типа (болезнь Херса). Редкий тип гликогеноза с изолированной гепатомегалией, являющейся следствием дефекта фосфорилазы печени. Наследование, вероятно, аутосомно-рецессивное. Наблюдается гепатомегалия без нарушения функции печени, отставание в росте. Биохимически — гипокалиемия, кетоз. Описываются клинические варианты с частичным блоком печеночной фосфорилазной активности (понижение активности на 10—15 и 25%). Прогноз этих форм благоприятный. У гетерозигот повышено содержание гликогена в эритроцитах. Патологоанатомически — накопление гликогена и жира в печени. Цирроз не развивается. Диагноз ставится на основании определения печеночной фосфорилазы в биоптате печени.
Гликогеноз VII типа (болезнь Томсона). Характеризуется накоплением фруктозо-6-фосфата, глюкозо-6-фосфата и глюкозы в мышцах, так как имеется дефицит фермента фосфоглюкомутазы. Клинические и морфологические изменения соответствуют мышечному гликогенозу V типа. После длительного мышечного напряжения — мышечная слабость, боли, напряженность. Ферментная активность снижена также в эритроцитах, что приводит к их гемолизу. Диагноз ставится но определению ферментов в биоптатах мышц.
Гликогеноз VIII типа (болезнь Таруи). Это мышечный гликогеноз с дефицитом фосфофруктокнназы. Клинически и морфологически сходен с гликогенозами V и VII типов.
Гликогеноз IX типа описан Hug G. и соавт. в 1966 г. Наследование по рецессивному типу, связанному с Х-хромосомой. Болезнь характеризуется дефектом фосфорилазы В в печени. Клиника и морфология сходны с гликогенозом VI типа.
X и XI типы гликогенозов характеризуются дефицитом ферментов протеинкиназы и фосфогексоизомеразы.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Гликогенозы. Классификация и проявления гликогеноза Гирке
Важнейшим признаком патологического накопления гликогена в тканях при гликогенозах является отсутствие постмортального гликолиза. При этом поглощенный гликоген можно легко экстрагировать водным раствором формалина. Раствор становится мутным, серовато-белого (молочного) цвета. При воздействии алкоголя из него выпадают студневидные массы, дающие с йодом выраженную бурую окраску. Однако не всякое наличие гликогена в тканях следует оценивать как гликогепоз.
Необходимо иметь в виду, что трупный гликолиз у плодов, новорожденных и грудных детей, особенно недоношенных, по сравнению с гликолизом у взрослых и детей старшего возраста очень замедлен в связи со слабой активностью их ферментных систем.
Гликогеноз I типа, гепаторенальный (болезнь Гирке), — наследственность аутосомно-рецессивная. Возникает на почве дефицита фермента глюкозо-6-фосфатазы. Гетерозиготные носители здоровы, однако их тромбоциты имеют повышенное содержание гликогена (тест для определения гетерозиготности).

Нарушения обмена выявляются в раннем грудном возрасте: отсутствие аппетита, отставание в физическом развитии, рвота. Постепенно развивается гипогликемия, периодически— кетонемические кризы. Отмечается пропорционально малый рост по гипофизарному типу, лицо имеет характерный кукольный вид, нарастает гепатомегалия. Можно пальпировать увеличенные почки. С возрастом развивается бледно-желтая окраска кожи.
Однако печеночная и почечная функции заметно не страдают, наблюдается отставание в физическом развитии. Прогноз неблагоприятный. Смерть наступает или от интеркуррентной инфекции, или от ацидотической комы. Описаны редкие случаи смерти от почечной недостаточности. Печень увеличена почти в 3 раза, поверхность ее гладкая, капсула напряжена, консистенция плотно-эластическая, край печени закруглен. Поверхность разреза бледно-красная, слегка пестрая, с подчеркнутым рисунком долек.
Содержание гликогена увеличено в 3—6 раз. Почки увеличены, набухшие, бледные, желтовато-красные, с широкой корковой зоной. Селезенка не увеличена. Микроскопически бросаются в глаза резко увеличенные, вздутые печеночные клетки с водянистой цитоплазмой, с отчетливо контурирующнмися границами и хорошо различимым центрально расположенным ядром. Окраска на гликоген резкоположительная. Одновременно определяется мелкокапсльнос, нерезко выраженное ожирение.
В проксимальных канальцах почек эпителий светлый, с подчеркнутыми границами, содержит большое количество гликогена (до 6,5%) (в норме по Эссбаху только 0,062% в свежей ткани). В надпочечниках атрофия преимущественно мозгового слоя.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Что такое Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) -
Гликогеноз третьего типа связан с мутациями структурного гена цитозольной амило-1,6-глюкозидазы, экспрессирующейся во многих тканях: печени, мышцах, эритроцитах. Ген картирован на хромосоме 1р21. Как и предыдущие варианты, третий тип гликогеноза наследуется по аутосомно-рецессивному типу.
Что провоцирует / Причины Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Гликогеноз III наследуется по аутосомно-рецессивному типу. Нередко родители состоят в кровном родстве. В одной семье иногда болеют брат и сестра или два брата. В основе заболевания лежит мутация гена, которая выявляется клинически у гомозиготов.
Патогенез (что происходит?) во время Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Патологическая анатомия. В печени, мышцах и сердце происходит накопление гликогена. При химическом исследовании обнаруживается аномалия структуры гликогена (лимитдекстрин). Гистологически выявляются большие набухшие фибриллы, подвергшиеся вакуолизации. Гепатоциты вакуолизированы и выглядят пенистыми, а в портальных пространствах отмечаются фиброз и круглоклеточная инфильтрация.
Электронно-микроскопически гликоген печени выявляется в виде альфа- и бета-частичек, клеточные органеллы нормальны и находятся вне скоплений гликогена. Клинически этот тип гликогеноза напоминает I тип, однако симптомы не столь резко выражены. Дети невысокого роста, с кукольным лицом и большим животом.
Отмечается увеличение подкожной жировой клетчатки на лице и на туловище, в связи с чем конечности выглядят тонкими. Важным клиническим симптомом является значительная гепатомегалия, которая отмечается уже на первом-втором месяце жизни. Печень быстро увеличивается и занимает - брюшной полости.
Симптомы Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Клинические проявления варьируют.
Различают две клинические формы с хроническим течением:
- IIIа- при которой возникают симптомы поражения печении мышц и I
- IIIb - при которой поражается только печень.
Диагностика Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Диагностика заболевания проводится на основе клинической картины и данных лабораторного исследования: снижения активности амило- 1,6-глюкозидазы и отложения гликогена измененной структуры в гепатоцитах и мышцах. В плазме крови отмечается увеличение концентрации лактата, мочевой кислоты, холестерина и триглицеридов.
Лечение Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Из-за нарушения гликогенолиза при гликогенозе типа III продукция глюкозы недостаточна, поэтому у грудных детей и детей младшего возраста после ночного голодания возникает гипогликемия. Усиление глюконеогенеза приводит к снижению уровня аминокислот в плазме (они используются как субстраты глюконеогенеза).
Таким образом, цель лечения - предупредить гипогликемию голодания и возместить дефицит аминокислот. Проводится оно следующим образом:
- прием необходимого количества глюкозы в виде сырого кукурузного крахмала в сочетании с диетой, содержащей достаточное количество белков и других питательных веществ, устраняет метаболические нарушения и задержку роста;
- больным с выраженной задержкой роста и тяжелой миопатией показано непрерывное ночное зондовое питание смесью, содержащей глюкозу, олигосахариды и аминокислоты, и частый прием богатой белком пищи в дневное время.
К каким докторам следует обращаться если у Вас Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Читайте также: