
Миелоидная метаплазия селезенки при сепсисе
Обновлено: 12.05.2024
Клиника хронического идиопатического миелофиброза (ХИМФ) - спленомегалия, портальная гипертензия, анемия, асцит
Картина крови при хроническом идиопатическом миелофиброзе (ХИМФ) отличается большим разнообразием. Показатели красной крови на момент постановки диагноза чаще нормальны или несколько снижены, но у 15—25 % больных уже имеется анемия. Повышение показателей красной крови, обычно клинически бессимптомное, отмечается у 5—6 % больных.
У большинства больных наблюдается нейтрофильный лейкоцитоз (10—15•10 9 /л), очень характерен выраженный палочкоядерный сдвиг и присутствие единичных мета- и миелоцитов.
У 10—15 % больных наблюдается более выраженный лейкоцитоз (>20•10 9 /л) и большая степень левого сдвига лейкоцитарной формулы крови, циркуляция единичных бластов. Число тромбоцитов нормально у 30—40 % больных и увеличено примерно у 15 %. Отмечается повышение уровня лактатдегидрогеназы, коррелирующее с числом лейкоцитов. Морфологические изменения эритроцитов — частая находка при хроническом идиопатическом миелофиброзе (ХИМФ). Клинические симптомы хронического идиопатического миелофиброза можно разделить на:
1) ассоциированные со значительным увеличением селезенки;
2) обусловленные усиленным клеточным катаболизмом;
3) обусловленные недостаточностью костного мозга.
Под последними имеются в виду анемический и тромбоцитопенический синдромы, хотя в действительности в их развитии участвуют разные патогенетические механизмы.
В зависимости от давности хронического идиопатического миелофиброза (ХИМФ) размеры селезенки широко варьируют. Со временем она достигает гигантских размеров, занимает всю левую и часть правой половины живота, отличается повышенной плотностью и бугристостью. Субъективные расстройства, вызываемые большой селезенкой, — это чувство тяжести, ощущение сдавления (малой вместимости) желудка и кишечника (неустойчивый стул), периодические острые боли, вызываемые инфарктом селезенки и периспленитом.
Классической причиной увеличения селезенки оказывается миелоидная метаплазия селезенки (ММС), но следующей возможной причиной спленомегалии является осложнение портальной гипертонией, а также увеличение депонирующей и секвестрирующей функций селезенки, т. е. рабочая гипертрофия органа.

Гепатомегалия и портальная гипертензия при хроническом идиопатическом миелофиброзе
Более чем у половины больных при установлении диагноза определяют гепатомегалию. Изолированное увеличение печени изредка возможно, как и преобладание гепатомегалии над спленомегалией. Значительное увеличение печени наблюдается обычно у больных, перенесших спленэктомию (СЭ), а частота прогрессирующей гепатомегалии после СЭ составляет 26—22 %.
Функциональные нарушения печени редки, чаще наблюдаются в терминальной стадии заболевания.
Развитие синдрома портальной гипертонии сопровождается значительным увеличением селезенки, не обусловленным ее участием в кроветворении, варикозным расширением вен пищевода, а затем периферическими отеками и асцитом. По данным Silverstein, портальная гипертония осложняет течение хронического идиопатического миелофиброза в 6—8 %, из них в 70 % она является результатом гиперкинетического тока крови и в 30 % — внутрипеченочных блоков. Их причины — миелоидная метаплазия, которая локализуется в синусоидах печени, вызываемый ею реактивный фиброз, а в отдельных случаях — постнекротический цирроз печени, обусловленный перенесенным гепатитом.
Среди вариантов портальной гипертонии, частота которой другими авторами оценивается в 10—15 %, выделяют пресинусоидальный тромботический блок, синусоидальную обструкцию, вызываемую миелоидной метаплазией печени в сочетании с гиперкинетическим током крови, постсинусоидальный, очевидно, тромботический блок, аналогичный синдрому Бадда — Киари.
Считается, что при функциональном внутрипеченочном портальном блоке анатомических изменений в печени нет, но есть и такая точка зрения, что для развития портальной гипертонии одного гиперкинетического тока крови недостаточно и структурные нарушения в печени должны присутствовать.
Уровень блока току крови в портальной системе в настоящее время определяется неинвазивным методом — ультразвуковой допплерографией.
При подпеченочной портальной гипертензии (ПГ) (тромбоз селезеночной или воротной вены) имеются большие размеры селезенки и отсутствие или незначительность увеличения печени. При пункции селезенки кровь поступает в шприц под большим давлением, в пунктате селезенки элементов миелоидной метаплазии обычно мало, за исключением случаев с большой продолжительностью заболевания и смешанным генезом спленомегалии.
При осложнении тромбозом надпеченочных вен клиническая картина выглядит значительно более драматично: болевой синдром имеет различную выраженность, но может и отсутствовать; формируется массивный, резистентный к лечению асцит, нередко желтуха, признаки печеночной недостаточности (клинические и лабораторные) тяжелое общее истощение; возможны периодические кровотечения из расширенных вен пищевода и желудка (кровавая рвота и мелена). Печень обычно значительно увеличена, тогда как селезенка — умеренно. Течение синдрома Бадда — Киари может быть острым, подострым и хроническим.
Неизвестно почему, но у всех шести наблюдаемых нами больных это осложнение возникло на раннем этапе, до постановки гематологического диагноза, и у женщин молодого возраста. Изменения в анализах крови были не столь однозначными, чтобы на их основании поставить гематологический диагноз и уточнить, какой именно. С подобной ситуацией сталкиваются и другие авторы. Расширение возможностей углубленного обследования больных с помощью мегакариоцитарной и эритроидной культур и цитогенетического анализа позволило прийти к заключению, что у 2/3 больных этот синдром является осложнением ХМПЗ.
Склонность к тромбозам в системе воротной, надпеченочных и чревных вен в целом отмечена преимущественно у больных эссенциальной тромбоцитемией (ЭТ) с моноклональным ге-мопоэзом, у носителей гена PRV-1, у больных со спонтанным ростом эритроидной культуры. Пока неизвестно, в какой степени эти патогенетические особенности распространяются на больных хроническим идиопатическим миелофиброзом (ХИМФ).
В клиническом отношении значение проблемы портальной гипертензии у больных хроническим идиопатическим миелофиброзом весьма существенно. Ее своевременная диагностика позволяет принять решение в пользу назначения спленэк-томии у больных с под- и внутрипеченочной портальной гипертензией и назначить адекватное консервативное или хирургическое (наложение шунтов) лечение при осложнении тромбозом надпеченочных вен.
Чаще всего большие размеры селезенки врачи относят за счет основного заболевания и проводят довольно агрессивную терапию с целью ее сокращения, что в случаях осложнения портальной гипертензией обречено на неудачу и может привести к серьезным проблемам.
В одном нашем наблюдении больную лечили по месту жительства миелосаном в течение 6 мес с целью сокращения размеров селезенки. В анализах крови наблюдались только небольшой тромбоцитоз и нейтрофилез. Заболевание расценивалось как сублейкемический миелоз. Селезенка была увеличена до уровня пупка. К концу этого лечения развился тяжелый геморрагический тромбоцитопенический синдром, в течение 3 мес больная находилась в критическом состоянии. Еще через 3 мес она была подвергнута спленэктомии. На операции выявлен цирроз печени без миелоидной метаплазии селезенки (ММС). Диагноз пересмотрен в пользу эссенциальной тромбоцитемии (ЭТ).
В течение последующих 5 лет больная находилась в хорошем состоянии и принимала гидроксимочевину в небольшой дозе, которая контролировала тромбоцитоз. Затем внезапно развился тромбоз в системе мезентериальных сосудов, распознанный с опозданием. Во время операции удалена значительная часть тонкого кишечника.
Случай демонстрирует часто имеющую место неточность гематологического диагноза в группе ХМПЗ, нераспознанную внутрипеченочную портальную гипертензию, в данном случае, видимо, обусловленную сопутствующим заболеванием, неадекватную цитостатическую терапию, осложнившуюся гипоплазией кроветворения и геморрагическим синдромом, а также развитие тромбоза мезентериальных сосудов при контролируемом тромбоцитозе.
Причиной развития асцита может оказаться не только портальная гипертензия, но и имплантация очагов кроветворения на брюшине и сальнике. В таких случаях в асцитической жидкости обнаруживают мегакариоциты и гранулоциты. Плевральный и абдоминальный выпот часто носит геморрагический характер. Эта и другие атипичные локализации миелоидной метаплазии: в лимфатических узлах со сдавлением спинного мозга, тонком кишечнике, средостении, почках, легких, других висцеральных органах — относятся к числу раритетов. Имеются данные об увеличении периферических лимфатических узлов у 32 % больных, но, по нашим наблюдениям, это значительно более редкий феномен.
К симптомам, обусловленным клеточным гиперкатаболизмом, относятся потеря массы тела и повышение температуры тела, гиперурикемия. Она может быть бессимптомной или протекать с признаками подагрической полиартралгии, подагры, мочекаменной болезни, осложняться хроническим пиелонефритом, обтурацией мочеточников, хронической почечной недостаточностью. У отдельных больных интенсивность камнеобразования в почках необычайно велика. Развитию урикемии способствует проведение массивной цитостатической терапии .
Хотя повышение температуры тела может быть результатом клеточного гиперкатаболизма, это справедливо по отношению к умеренному субфебрилитету, а значительные подъемы температуры тела обычно обусловлены инфекцией, особенно мочевыводящих путей, или латентно протекающим МДС-синдромом, который может проявиться как типичный, развернутый острый лейкоз через ряд месяцев и даже лет.

В случаях, протекающих с количественной и качественной патологией тромбоцитов, возможны сосудистые осложнения: тромботические микроциркуляторные расстройства, тромбозы артерий и вен, геморрагический синдром, ДВС-синдром.
Внутренние кровотечения обычно обусловлены разрывом вен пищевода при осложнении портальной гипертонией. Присущая этому заболеванию, как и другим ХМПЗ, качественная дефектность тромбоцитов объясняет появление экхимозов на коже при сравнительно умеренной тромбоцитопении. Несостоятельность гемостаза особенно четко проявляет себя при спленэктомии.
Анемический синдром нередко выходит на передний план, особенно в поздних стадиях заболевания. Его причины разнообразны. Среди них могут быть:
• недостаточное образование эритроцитов;
• гиперволемия;
• усиление депонирования и секвестрации клеток крови в увеличенной селезенке (гиперспле-низм);
• аутоиммунный гемолиз эритроцитов;
• ускоренный гемолиз эритроцитов в результате синдрома пароксизмальной ночной гемоглобинурии или ферментных дефектов (дефицит Г-6-ФДГ и др.);
• дефицит железа и фолиевой кислоты.
Количественная недостаточность эритропоэза определяется замещением кроветворного костного мозга миелофиброзом и остеомиелосклерозом с возможным присутствием жировой ткани. Компенсаторный эритропоэз в трубчатых костях со временем также редуцируется, а компенсаторные возможности селезеночного эритропоэза ограничены его частой неэффективностью и одновременным усилением депонирования и деструкции клеток крови в большой селезенке.
Гемодилюционная анемия является результатом гиперволемии, обусловленной спленомегалией. Она хорошо переносится больными и является по существу только лабораторным феноменом.
Дефекты мембраны эритроцитов, сходные с наблюдаемыми при пароксизмальной ночной гемоглобинурии (ПНГ), описаны при хроническом идиопатическом миелофиброзе многими авторами. Их последствием является синдром гемолитической анемии. Повышенному гемолизу эритроцитов способствует и усиление перекисного окисления липидов клеточных мембран эритроцитов.
Дефицит фолиевой кислоты, приводящий к появлению макроцитарной анемии с кольцами Кебота, базофильной пунктацией эритроцитов, тельцами Жолли, наблюдается в поздней стадии заболевания. Его объясняют повышенным расходом фолиевой кислоты при усиленном гемопоэзе.
К количественной недостаточности эритропоэза приводит и его подавление при прогрессирующей гиперплазии лейкоцитарного ростка, хронической и острой. Возможно развитие сидеробластной анемии без- и с малопроцентной бластемией, которая является предстадией острого лейкоза. Описаны случаи парциальной красноклеточной аплазии, завершившиеся острым лейкозом. Отметим, что обычно к анемии приводит сочетание нескольких причинных факторов. Удельный вес каждого из них подлежит уточнению, что особенно необходимо при решении вопроса о назначении спленэктомии. Это же относится и к тромбоцитопеническому синдрому. Причинами его развития являются:
• усиление депонирования и деструкции тромбоцитов в увеличенной селезенке (и печени);
• вторичный аутоиммунный гемолиз тромбоцитов;
• нарушение образования тромбоцитов в результате редукции числа мегакариоцитов или их качественной дефектности;
• сочетание этих процессов;
• ДВС-синдром (тромбоцитопения потребления).
Среди известных клинических проявлений заболевания могут иметь место и аутоимунные симптомы, такие как дерматиты и кожные васкулиты, опосредованные активацией Т-лимфоцитов.
При рентгенографическом исследовании костного скелета обнаруживаются признаки уплотнения структуры плоских костей, особенно позвонков, иногда эбурнеация трубчатых костей с сужением их просвета, иногда определяют очаговый остеолиз.
Эволюция хронического идиопатического миелофиброза характеризуется постепенным нарастанием лейкоцитоза при исходно различном числе лейкоцитов: нормальном, сниженном и повышенном. Развитие острого лейкоза наблюдается как в случаях прогрессирующего лейкоцитоза (более 30•109/л), так и лейкопении (3•109/л). Большинство больных не доживает до развития типичного острого лейкоза. Картины рефрактерной анемии, тромбоцитопении, панцитопении или, наоборот, нарастающего лейкоцитоза и левого сдвига лейкоцитарной формулы, выход из-под контроля размеров селезенки, появление упорной асептической лихорадки являются показателями терминальной фазы заболевания.
Терминальное состояние больных может определяться и висцеральными осложнениями: сердечной, печеночной и почечной недостаточностью, к которым имеются соответствующие патофизиологические предпосылки. Жизнь больных может оборвать острое кровотечение из расширенных вен пищевода при осложнении портальной гипертонией. Гематологические и соматические терминальные состояния нередко сочетаются с тяжелой общей дистрофией и компрессионными осложнениями.
Возможны варианты более злокачественного течения, которые терминологически обозначаются как варианты с ускоренным развитием терминальной фазы. Выделяют и вариант с подострым течением, при котором морфологические изменения костного мозга не отличаются от обычного хронического идиопатического миелофиброза, но при этом нет значительной спленомегалии, и довольно быстро развиваются анемия и другие проявления недостаточности кроветворения.
К атипичным хронически протекающим вариантам хронического идиопатического миелофиброза можно отнести и так называемые гибридные формы, имеющие признаки двух ХМПЗ:
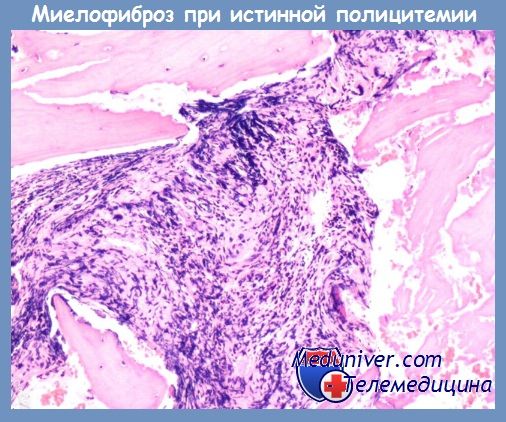
• истинной полицитемии и хронического идиопатического миелофиброза — упорная плетора, но раннее и значительное увеличение селезенки за счет миелоидной метаплазии, лейкоцитозные формы, устойчивость к цитостатической терапии, эволюция в острый лейкоз через длительный период зрелоклеточного лейкоцитоза, частые цитогенетические аномалии;
• хронического идиопатического миелофиброза и эссенциальная тромбоцитемия. С первым заболеванием их сближает выраженный миелофиброз, со вторым — небольшая величина селезенки, значительный тромбоцитоз, отсутствие лейкоэритробластической картины периферической крови, характерной для иМФ.
Атипичными в определенном смысле являются и случаи хронического идиопатического миелофиброза с существенным увеличением печени, а не селезенки, а также осложненные портальной гипертонией, синдромом Бадда — Киари, при которых проявления гематологического заболевания по анализу крови могут быть минимальными, и только культуральные и цитогенетические исследования выявляют их принадлежность к ХМПЗ.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Течение истинной полицитемии - гематологические исходы
Наиболее часто истинная полицитемия завершается развитием миелофиброза с миелоидной метаплазией (МММ) селезенки. Частота этого исхода составляет 15—20 %, но у проживших более 20 лет она увеличивается до 30 % и более. МММ завершает эритремическую стадию истинной полицитемии. Это естественная эволюция заболевания для всех больных, если они не умирают от тромбозов сосудов, острого лейкоза, других неоплазий, сопутствующих заболеваний и висцеральных осложнений. Продолжительность эритремической стадии в среднем составляет 10—15 лет, но не являются редкостью и более короткие, и более продолжительные ее сроки (в отдельных собственных наблюдениях до 20—25 лет).
Патоморфодинамика этого исхода хорошо изучена с помощью прижизненной трепанобиопсии подвздошной кости и пункции селезенки, патофизиология — с помощью радиологических методов исследования.
Миелоидная метаплазия селезенки (ММС) развивается еще в эритремической (2Б) стадии заболевания и со временем приводит к значительной сплено-мегалии. Обычно она носит трехростковый характер с преобладанием эритропоэза. ММС по времени возникновения опережает развитие миелофиброза.
Развитию коллагенового, определяемого при окраске гематоксилин-эозином миелофиброза предшествует ретикулиновый, для обнаружения которого используются методы импрегнации серебром по Футу и Гомори. На стадии ретикулинового миелофиброза клеточный костный мозг еще весьма гиперплазирован. Коллагеновый миелофиброз вначале сосуществует с клеточной пролиферацией, а потом подавляет ее. Дополнительное развитие остеомиелосклероза наблюдается реже, чем при ХИМФ. Патоморфологическая картина этого периода напоминает изменения костного мозга, наблюдаемые при ХИМФ в 3—4-й стадии его развития.
Это относится и к резко измененному состоянию микро- и макрососудов, мелких артерий и артериол, синусов и синусоидов стромы костного мозга.
При миелофиброзе с миелоидной метаплазией (МММ), развивающемся после истинной полицитемии, значительно меняется топография гемопоэза, что имеет диагностическое значение. Наблюдается падение захвата коллоидного 99mТс осевым скелетом и усиление захвата изотопа эпифизами трубчатых костей, селезенкой и печенью. Возможны различные варианты в степени этих изменений.
Развитие постэритремического миелофиброза происходит под влиянием фиброгенных цитокинов, главным источником которых являются патологические мегакариоциты, тромбоциты и моноциты.
Общность происхождения постполицитемического и ХИМФ отражают и данные цитогенетического исследования.

По данным A. Tefferi и соавт., частота нарушений кариотипа составила 62 % на момент первого исследования, 73 % в динамике заболевания, 90—100 % в период развития острого лейкоза. Характер выявленной цитогенетическои патологии при постполицитемическом МММ был таким же, как и при ХИМФ: 13q-, 20q-, +8, +9. В динамике любого МММ частота 13q- и 20q- значительно возрастает.
Авторы пришли к выводу, что 13q-, 20q- и +8 цитогенетическая патология не имеет плохого прогностического значения. Появление другой цитогенетическои патологии и новых субклонов обычно индуцировано предшествующей цитостатической терапией и имеет плохое прогностическое значение: определены связь между частотой развития острого лейкоза при МММ и обнаружением 5q- в 50 %, 7q- - в 30 %, 12р- — в 25 %, нарушения в хромосоме 1 — в 25 %. Случаев развития острого лейкоза у имевших 13q- не обнаружено.
Клиническая и гематологическая динамика процесса развития миелофиброза с миелоидной метаплазией (МММ) характеризуется прогрессирующим увеличением селезенки, а часто и печени (в единичных случаях гепатомегалия выходит на передний план), потерей массы тела вплоть до кахексии, нередко — появлением субфебрилитета и более высоких подъемов температуры телы у отдельных больных, повышением частоты инфекционных осложнений, проявлениями уратового диатеза, портальной гипертонии, сохранением склонности к тромбозам сосудов и экхимозам.
Большая величина селезенки определяет симптом компрессии желудка и дисфункцию кишечника, смещение левой почки. Возникновение острых болей в области селезенки определяется инфарктами селезенки с возможным развитием периспленита.
Показатели красной крови в процессе развития миелофиброза с миелоидной метаплазией (МММ) постепенно нормализуются, а затем возникает анемия с морфологическими изменениями в эритроцитах, присущими ХИМФ: пойкилоцитоз с грушевидной формой эритроцитов, анизоцитоз и нормобластемия. Характерно нарастание числа лейкоцитов и левого сдвига в лейкоцитарной формуле крови; у отдельных больных развивается лейкопения. Число тромбоцитов варьирует от высоких до низких значений.
На поздних этапах миелофиброза с миелоидной метаплазией (МММ) нередко возникает макроцитарная анемия с токсигенной зернистостью в эритроцитах, шизоцитами и кольцами Кэбота. Причиной ее развития является относительный дефицит фолиевой кислоты, а не витамина В12.
Основные причины развития анемии при постполицитемическом МММ — повышенный гемолиз эритроцитов в резко увеличенной селезенке, неэффективный эритропоэз с гибелью части эритроидных предшественников и коммитированных клеток в костном мозге, доказанной радиологическими исследованиями, подавление эритропоэза диффузным миелофиброзом и лейкемизированным миелопоэзом. К ним добавляются такие факторы, как гидремия, дефицит железа, оставшийся от эритремической стадии, относительный дефицит фолиевой кислоты. Отдельные причины часто сочетаются у одного и того же больного.
В зависимости от причин развития анемический синдром имеет различную тяжесть и прогноз от вполне благоприятного при железодефицитной до тяжелого при рефрактерной анемии и миелодисплазии, одним из вариантов которой является сидеробластная анемия.
Течение постэритремической миелоидной метаплазии селезенки и миелофиброза необычайно вариабельно. У некоторых больных оно вполне доброкачественное с медленными темпами роста размеров селезенки и печени, нормальными показателями красной крови, умеренным лейкоцитозом и тромбоцитозом или без них. Подобную многолетнюю благополучную динамику заболевания можно обозначить как период стабилизации. У других больных отмечается быстрое прогрессирование спленомегалии, анемический синдром, нарастающий лейкоцитоз с малопроцентной бластемией.
Эти варианты лейкоцитоза, зрелоклеточного или напоминающего хронический миелолейкоз, с высокой вероятностью предвещают развитие миелодисплазии или острого лейкоза. Это стандартный исход истинной полицитемии, протекающей с лейкоцитозом выше 30•109/л.
Плохое прогностическое значение имеет и лейкопения меньше 3•10 9 /л, особенно если она сочетается с выраженной анемией и омоложением лейкоцитарной формулы. Подобного рода состояние, как и лейкоцитозные формы при наличии рефрактерной к лечению анемии, сопровождающееся повышением температуры тела и/или появлением геморрагического синдрома, рассматривается как миелодиспла-зия, предстадия острого лейкоза. Так же трактуются и случаи сидеробластной анемии. В этих случаях можно ожидать прогностически неблагоприятных цитогенетических находок.
Итальянские авторы рассматривают всю стадию постполицитемического МММ как предостролейкозную с вероятностью развития острого лейкоза через 3 года после ее диагностики. Если с первым положением условно можно согласиться, то это не относится к обозначенному авторами сроку, поскольку у многих больных он достаточно высокий, а часть больных и не доживает до его развития.
Манифестации острого лейкоза предшествует помимо рефрактерной анемии и нарастающего лейкоцитоза ряд признаков:
• асептическая лихорадка, продолжительность которой до диагностики острого лейкоза может составить 1—2 года;
• глубокая цитопения, в том числе и тромбоцитопения, немотивированная большой величиной селезенки и предшествующей цитостатической терапией;
• быстрый рост размеров селезенки, особенно если он сочетается с лихорадочным синдромом.
• тромбоцитемия на позднем этапе заболевания, если ее раньше не было (2 собственных наблюдения).
Манифестации острого лейкоза могут предшествовать трудно квалифицируемые дерматиты лица, глосситы, резистентность к прежде адекватному лечению. Подозрение на острый лейкоз возникает и в случаях захвата осевым скелетом 99mТс при наличии выраженного миелофиброза в гистоморфологическом препарате.
Развитие острого лейкоза возможно как в эритремической стадии заболевания, так и в стадии постэритремической ММС и миелофиброза. В последнем случае преобладают лейкоцитозные варианты с частичным вызреванием, в крови встречаются эритрокариоциты, осколки ядер мегакариоцитов. Чаще всего имеет место миелобластный вариант острого лейкоза, но возможны и наблюдались нами эритромиелоз, миеломоноцитарный и лимфобластный варианты. Для клинических проявлений острого лейкоза во всех случаях были характерны мучительные оссалгии.
К настоящему времени уже нет сомнений в том, что естественные тенденции к завершению заболевания острым лейкозом при истинной полицитемии имеются, но они выражены незначительно. У леченных цитостатическими препаратами частота развития острого лейкоза существенно возрастает, как и заболеваемость раком и злокачественными лимфомами.
По мере получения доказательств лейкозогенности цитостатической терапии стимулировались поиски новых методов лечения.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Патологоанатомическая диагностика сепсиса : методические рекомендации / О.Д.Мишнёв, А.И.Щёголев, О.А.Трусов. — Москва, 2004.
библиографическое описание:
Патологоанатомическая диагностика сепсиса : методические рекомендации / Мишнёв О.Д., Щёголев А.И., Трусов О.А. — 2004.
код для вставки на форум:
Российское общество патологоанатомов
О.Д.Мишнев, А.И.Щеголев, О.А.Трусов
ПАТОЛОГОАНАТОМИЧЕСКАЯ ДИАГНОСТИКА СЕПСИСА
Введение
Сепсис – полиэтиологическое инфекционное заболевание, характеризующееся генерализованным характером, ацикличностью течения и особым образом измененной реактивностью. Сепсис развивается из местных очагов инфекции и рассматривается, как правило, в качестве осложнения заболеваний, послуживших причиной генерализации инфекции. Последние годы, несмотря на интенсивную терапию, характеризуются ростом числа больных сепсисом и высокой летальностью.
Сепсис представляет собой заболевание, в отношении этиологии, патогенеза, патологической анатомии, диагностики, лечения которого всегда были и ещё будут проводиться дискуссии. Клиницисты обращаются к патологоанатомам в надежде получить желаемое и в то же время, по их мнению, объективное и адекватное объяснение летального исхода при сепсисе. Однако, справедливости ради, следует констатировать, что, как правило, такое объяснение для них a priori, до вскрытия, в современных клиниках уже имеется, и оно может быть получено на основании комплекса клинических и лабораторных признаков доказательной медицины.
Именно эти показатели, очевидно, следует использовать и патологоанатому при анализе истории болезни и результатов вскрытия для решения своих диагностических проблем. Многие клиницисты интересуются на вскрытии, т.е. a posteriori, не столько познанием на морфологическом субстрате причины и действия, сколько прагматичным согласованием формальных параметров, определяемых при сличении клинического и патологоанатомического диагнозов. Это определяет первоочередную задачу совместной междисциплинарной работы – нахождения общих точек соприкосновения клиницистов и патологоанатомов для полноценного и современного анализа результатов патологоанатомического исследования умерших от сепсиса.
Теоретические аспекты проблемы сепсиса, представленные в мировой литературе конца ХХ – начала нынешнего века и воспринимаемые как революционные и во многом совершенные, дающие основания для разработки оптимальных методов лечения и недопущения летальных исходов при сепсисе, не были неожиданными для патологоанатомов России. В этом заслуга наших Учителей и в первую очередь И.В. Давыдовского, а также А.В.Смольянникова, Д.С.Саркисова, А.П.Авцына, Н.К.Пермякова и других, которые первоочередное внимание уделяли макробиологической составляющей сепсиса, не забывая при этом об этиопатогенетической роли возбудителей инфекции.
Принятие и выполнение решений Согласительной конференции пульмонологов и специалистов по интенсивной терапии (ACCP/SCCV), состоявшейся в 1991 году в Чикаго, привело к унификации терминологии и к упрощению клинической диагностики сепсиса. Однако отсутствие рандомизированных патологоанатомических исследований и, соответственно, клинико-морфологических сопоставлений является одной из важных причин неполного взаимопонимания клиницистов и патологоанатомов при оценке формы и танатогенеза заболевания, а также при обсуждении расхождений клинического и патологоанатомического диагнозов. К сожалению, до настоящего времени предметом дискуссий является отнесение тех или иных клинических и морфологических признаков к проявлениям сепсиса.
Также следует остановиться на распространённой ныне тенденции считать условным выделение разновидностей сепсиса по входным воротам, поскольку генерализованная реакция организма на инфекцию по своей сути является единой и требует незамедлительного лечения, а в Международной классификации болезней отсутствует подразделение сепсиса на разновидности по данному основанию. Возможно, что такой прагматичный подход является обоснованным, однако мы рекомендуем патологоанатомам не забывать о морфологических особенностях различных форм сепсиса в зависимости от входных ворот.
Действительно, для патологоанатома и клинициста не будет представлять большой сложности достижение взаимопонимания при анализе результатов секции больного, погибшего от сепсиса при наличии типичных морфологических признаков и соответствующих клинических данных. Гораздо большие трудности возникнут в том случае, когда больного лечили от сепсиса (и по результатам вскрытия довольно успешно – именно так!), а смерть наступила от другой причины, видимо не связанной с сепсисом. В этом случае необходимо учитывать не только медицинские составляющие (организационные, научно–практические), но также социальные и экономические вопросы, которые в настоящее время никогда не следует упускать из вида. Также сложными для патологоанатомической диагностики являются наблюдения лечённого сепсиса в плане танатогенеза и установления непосредственной причины смерти. Наконец, наблюдения посмертно диагностированного сепсиса, который не был выявлен в клинике, должны стать предметом обсуждения на клинико–патологоанатомических конференциях.
Определения терминов и понятий
Сепсис (в переводе с греческого sēpsis – гниение) – представляет собой особую форму тяжелой генерализованной инфекции, при которой макроорганизм не способен локализовать инфекционный процесс (Белянин В.Л., Рыбакова М.Г., 2004).
Бактериемия – симптом, обозначающий наличие в крови живых микроорганизмов (не обязательно только при сепсисе).
Диссеминация – употребляется, как правило, для характеристики распространения поражений при инфекционном процессе в пределах одного органа (например, диссеминированный туберкулез легких).
Генерализация – обозначает, как правило, поражение многих органов и систем (например, генерализованная вирусная, туберкулезная инфекция).
Синдром системного воспалительного ответа (ССВО) (Systemic Inflammatory Response Syndrome (SIRS), синдром системной воспалительной реакции (ССВР)) патологическое состояние, обусловленное одной из форм хирургической инфекции или альтерации ткани неинфекционной природы (травма, панкреатит, ожог, ишемия или аутоиммунные повреждения тканей и др.). Это понятие предложено классификацией ACCP/SCCV, что привело к существенному пересмотру концептуальных положений о патогенезе, клинике, лечении, профилактике возникновения сепсиса и его осложнений. ССВР (SIRS) характеризуется наличием более чем одного из четырёх следующих основных клинических признаков: гипертермия, тахикардия, тахипноэ, лейкоцитоз (или лейкопения). Подобные клинические признаки встречаются при сепсисе, но при этом обязательным является наличие инфекционного очага поражения в тканях или органах. Патологоанатомическая характеристика SIRS является объектом перспективных научных исследований.
Септицемия – клинико–патологоанатомическая форма сепсиса, при которой отсутствуют метастатические септические очаги. Септицемия – это сепсис без гнойных метастазов. По поводу правомочности применения этого понятия в качестве диагноза одной из форм сепсиса до сих пор продолжается дискуссия. Некоторые клиницисты и патологоанатомы советуют вообще отказаться от этого термина (Пермяков Н.К., 1992). Этот термин не представлен и в классификации ACCP/SCCV. Тем не менее, он рекомендован МКБ-10 как унифицированный для всех разновидностей сепсиса, и вплоть до нового пересмотра он должен применяться клиницистами и патологоанатомами при определении шифра большинства клинико–патологоанатомических форм сепсиса.
Септикопиемия – клинико–патологоанатомическая форма сепсиса. Для неё характерно наличие гнойного воспаления (абсцессов, апостем, флегмон, интерстициального гнойного воспаления) в различных органах и тканях, возникающего в результате гематогенного распространения микробных эмболов из септического очага. Септикопиемия – это сепсис с гнойными метастазами.
Термин септикопиемия не представлен в МКБ–10.
Термин сепсис носит обобщающий характер, как и термин септический процесс. Классификация ACCP/SCCV рекомендует в клинике использовать термин сепсис, а не септицемия или септикопиемия. В настоящее время не рекомендуется использовать термин септическое состояние, лишенный конкретной основы.
Тяжёлый сепсис (или сепсис-синдром) – форма сепсиса, предложенная в классификации ACCP/SCCV, при котором отмечаются признаки полиорганной недостаточности. В.Л.Белянин и М.Г.Рыбакова (2004) считают, что с точки зрения патоморфолога понятие тяжёлый сепсис (сепсис–синдром) нельзя признать удачной терминологической находкой.
Септический шок – другая форма тяжёлого сепсиса в классификации ACCP/SCCV, сопровождающаяся артериальной гипотонией, не устраняющейся с помощью инфузионной терапии и требующей назначения катехоламинов.
Септический (бактериальный, инфекционный) эндокардит (острый и подострый) характеризуется, прежде всего, воспалительными поражениями клапанного аппарата сердца, развивающимися наряду с септическими проявлениями. Представляет собой самостоятельное заболевание (первоначальную причину смерти) и имеет собственный шифр. Следует отличать от поражений эндокарда при других формах сепсиса.
Септический очаг – различают первичный и метастатические септические очаги. Первичный септический очаг представляет собой фокус воспаления, преимущественно гнойного, в котором происходит накопление микроорганизмов, распространяющихся затем гематогенно и лимфогенно по тканям и органам. Метастатические септические очаги (вторичные септические очаги, метастатические очаги при сепсисе, септические метастазы, метастатические гнойники, гнойные метастазы) представляют собой очаги воспаления, возникающие вследствие гематогенного перемещения септического процесса в ткани и органы из другого очага.
Системный инфекционный процесс – поражение какой-либо системы органов (например: нервной при клещевом энцефалите; пищеварительной при шигеллезе и т.д.).
Гнойно-резорбтивная лихорадка. Понятие гнойно-резорбтивной лихорадки предложено И.В.Давыдовским для обозначения клинических и патологоанатомических проявлений местного гнойно-некротического процесса, морфологически ограниченного демаркационным валом (в отличие от септических очагов) от окружающих тканей. В настоящее время этот термин почти не используется, а состояние больного обозначается как интоксикация или разновидность эндотоксикоза.
Эндотоксикоз – очень широкое понятие для обозначения осложнений и проявлений заболеваний и состояний организма. Основным фактором патогенеза эндотоксикоза является воздействие на организм токсических продуктов, образующихся в нем в результате нарушения тех или иных функций.
Этиология
Возбудителями сепсиса могут являться многие микроорганизмы (табл. 1). Это многочисленные бактерии: стрептококки, стафилококки, менингококки, пневмококки, кишечная палочка, синегнойная палочка, клебсиеллы, протей, сальмонеллы, а также грибы преимущественно Кандида и аспергиллус. Сепсисом может осложниться течение туберкулеза, сыпного тифа, брюшного тифа и других инфекционных заболеваний, возбудителями которых являются патогенные микроорганизмы. Генерализованные вирусные инфекции не принято рассматривать в качестве сепсиса, хотя об этом имеются указания в современных зарубежных клинических исследованиях.
Таблица. 1. Наиболее вероятная этиология сепсиса в зависимости от локализации первичного очага инфекции
Лечение при постполицитемическом миелофиброзе с миелоидной метаплазией (МММ) - эффективность
Лечение в постэритремической стадии истинной полицитемии (ИП) определяется характером гематологического исхода. В целом это миелофиброз с миелоидной метаплазией (МММ), но с различной величиной селезенки, различием гематологических данных и гистоморфологии костного мозга — от гиперклеточного до диффузно-фиброзного.
Принципы лечения в стадии постэритремического миелофиброза с миелоидной метаплазией (МММ) аналогичны таковым при ХИМФ, с тем отличием, что при первом могут сохраняться повышенные показатели красной крови и, следовательно, необходимость в эксфузиях крови. Хотя опасность лейкозогенного действия цитостатических препаратов в этой стадии резко возрастает, их приходится назначать для контроля над прогрессирующим лейкоцитозом, нарастающим числом промежуточных форм белого ряда и спленомегалией. Большого опыта терапии ИФН-а этой стадии истинной полицитемии (ИП) еще нет, и вряд ли он будет очень успешным.
Следует отметить, что клинические и гематологические проявления миелофиброза с миелоидной метаплазией (МММ) неоднозначны. У целого ряда больных темпы роста селезенки весьма умеренны, анемии нет и предметом коррекции оказывается один тромбоцитоз. Возможно, что для таких больных средством выбора будет анагрелид, но нами применялась Hydrea в течение длительного срока с хорошей переносимостью и эффектом.
По опыту отечественной гематологии, лейкозогенное действие Hydrea несравненно меньшее, чем у алкилирующих агентов, назначения которых в этой стадии действительно лучше избегать.
Для больных, завершивших эритремическую стадию периодом стабилизации, активная терапия может и вовсе не потребоваться.
Терапевтическая тактика в случаях прогрессирующего увеличения селезенки при отсутствии лейкоцитоза и ромбоцитоза проблематична. Попытки применения Hydrea в небольших дозах приносят малый эффект. Терапевтические возможности ИФН-а в подобной ситуации пока не определены. Нужно помнить, что прогрессирование спленомегалии может быть обусловлено не только нарастанием экстрамедуллярного кроветворения, но и внутрипеченочной портальной гипертонией (ПГ), обычно без больших анатомических предпосылок для этого.
Это так называемый функциональный блок в печени, что подтверждают результаты секционного исследования печени у умерших от пищеводных кровотечений. Больные с данным осложнением подлежат спленэктомии (СЭ), которая, однако, не гарантирует дальнейшего гематологического благополучия.
Лечение анемического синдрома в этой стадии осуществляется с учетом патогенеза анемии. Оно тождественно его лечению при ХИМФ, включая назначение спленэктомии (СЭ) при гемолитическом характере анемии.
Лечение анемии, вызванной нарушенным образованием эритроцитов в результате замещения кроветворного костного мозга диффузным миелофибро-зом, или неэффективного эритропоэза, или смены ведущей эритроидной линии пролиферации на гра-нулоцитарную, является наиболее трудной задачей. Это в полной мере относится и к рефрактерной си-деробластной анемии, предстадии острого лейкоза. Любая из указанных выше предпосылок к развитию анемии, в сущности, относится к числу терминальных состояний и плохо поддается коррекции, в том числе андрогенами и цитостатиками, глюкокортико-стероидами и ИФН-а.
Попытки лечения макроцитарных анемий фолиевой кислотой нами неоднократно предпринимались, но почти всегда безуспешно. Назначение препаратов железа при оставшемся от плеторической стадии его дефиците оправдано, если дефицит железа подтвержден исследованием уровня феррити-на. Показатели транспортного фонда железа часто дают ошибочные результаты.
Лечение тромбоцитопении также определяется причиной ее развития. Как и при ХИМФ, она является показанием к спленэктомии, если обусловлена гиперспленизмом. К тромбоцитопении может привести предшествующая цитостатическая терапия, и особенно терапия миелосаном. В подобных случаях спленэктомия не показана. Тромбоцитопения при малом числе мегакариоцитов в костном мозге и при преобладании мелких и атипичных по структуре форм чаще всего является проявлением миелодисплазии. В этих случаях спленэктомия неэффективна и не должна назначаться.
Опыт спленэктомии центра РАМН включает 12 больных. Повышенной кровоточивостью сопровождались практически все спленэктомии. Осложнения тромбофлебитами наблюдались у 2/3 пациентов почти у всех имелся тромбоцитоз в послеоперационном периоде. Двое больных погибли от осложнений, связанных с операцией. Причины смерти 2 больных — миелодисплазия, у 3 — острый лейкоз.
Продолжительность жизни у перенесших спленэктомию больных составила 1,5 года — 8 лет.
При анализе отдаленных результатов спленэктомии у нас не сложилось впечатления, что операция явилась причиной развития острого лейкоза, скорее, она не предотвратила его развития. Целесообразность спленэктомии в случаях миелодисплазии крайне сомнительна; когда же больные оперируются по поводу компрессионного синдрома при гематологической стабильности, портальной гипертонии, гемолитической анемии, достигаются хорошие результаты.
В постэритремической стадии возрастает необходимость в симптоматическом лечении. Объектами этой терапии становятся истощение больного, при котором показаны анаболические гормоны, гиперкатаболический синдром, когда полезными оказываются кортикостероиды (преднизолон в суточной дозе 10—15 мг), уратовый диатез, при котором необходима терапия аллопуринолом, и сосудистые тромбофилические и геморрагические осложнения. Для симптоматической терапии массивной спленомегалии (в случаях, когда спленэктомия противопоказана) применяется гамма-терапия.
Прежде она назначалась часто, но получаемые результаты были кратковременными (в среднем 7—8 месяцев), цитопенические осложнения — нередкими, темпы роста селезенки после ее временного сокращения — возрастающими. Это объясняет общее крайне сдержанное отношение к данному методу лечения.
Лечение острого лейкоза у больных истинной полицитемией (ИП) проводится по общим правилам и иногда успешно.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Миелофиброз – это хроническое гематологическое заболевание, характеризующееся опухолевой пролиферацией гемопоэтических стволовых клеток и фиброзом костного мозга. Основные клинические проявления включают симптомы опухолевой интоксикации и анемического синдрома (прогрессирующую слабость, бледность кожи и слизистых оболочек, потерю веса), а также увеличение селезенки (спленомегалию). Диагноз устанавливается на основании молекулярно-генетических исследований, изучения гистологической картины костного мозга. Лечение проводится с помощью химиотерапевтических препаратов. Хирургические методы лечения подразумевают трансплантацию костного мозга и удаление селезенки.
МКБ-10

Общие сведения
Миелофиброз (агногенная миелоидная метаплазия, сублейкемический миелоз) – злокачественное заболевание, при котором происходит постепенное замещение костного мозга опухолевыми стволовыми клетками и разрастающейся соединительной тканью. Впервые эту патологию описал немецкий врач Г. Хойк в 1879 году. А в 1951 году американским гематологом Уильямом Дамешеком миелофиброз был выделен в самостоятельную нозологическую единицу. При неблагоприятном течении миелофиброз способен трансформироваться в более тяжелую болезнь ‒ острый лейкоз. Распространенность миелофиброза составляет от 0,3 до 0,7 случаев на 100 тыс. населения. Пик заболеваемости приходится на возраст от 50 до 70 лет, но встречаются и молодые пациенты. Чаще страдают мужчины.
Причины миелофиброза
Существует первичный и вторичный сублейкемический миелоз. Точная причина первичного миелофиброза до сих пор не установлена. Наибольшей популярностью среди специалистов в области гематологии пользуется теория влияния генетической мутации. У большинства пациентов выявляются мутации гена тирозинкиназы (JAK2V617F), кальретикулина (CALR), тромбопоэтина (MPL), регулирующих экспрессию белков JAK-STAT сигнального пути. Гены локализуются в локусе хромосомы del3p24.
В качестве этиологического фактора изучается действие большой дозы радиоактивного излучения. Также рассматривается роль персистирующих вирусных инфекций (вируса простого герпеса, Эпштейна-Барра, цитомегаловируса), длительного приема оральных контрацептивов, миелосупрессивных лекарственных препаратов, контакта с различными органическими и неорганическими соединениями (бензолом, мышьяком). Вторичный миелофиброз развивается как исход других хронических миелопролиферативных заболеваний – истинной полицитемии, эссенциальной тромбоцитемии, хронического миелолейкоза.
Патогенез
В результате повышенной экспрессии сигнальных белков в одной из стволовых костномозговых клеток запускается активная пролиферация (опухолевая трансформация). Этот процесс сопровождается вторичным воспалением с выделением цитокинов и факторов роста. Факторы роста фибробластов и эндотелия сосудов индуцируют выработку стромальными клетками костного мозга большого количества коллагена и разрастание соединительной ткани (собственно фиброз). Постепенно нормальная ткань костного мозга замещается опухолью и соединительной тканью.
При массивном поражении опухолью костного мозга клетки крови, не достигнув стадии полного созревания, попадают в системный кровоток. Это приводит к образованию очагов экстрамедуллярного (внекостномозгового) кроветворения, главным образом в печени и селезенке. Распад опухоли ведет к высвобождению мочевой кислоты, которая откладывается в тканях суставов и почечных канальцах.
Симптомы миелофиброза
Длительное время пациент чувствует себя удовлетворительно. Через несколько лет от начала заболевания постепенно появляется опухолевая интоксикация в виде общей слабости, повышения температуры до субфебрильных цифр, потливости, усиливающейся по ночам. У больного снижается аппетит, он стремительно теряет в весе. Присоединяется анемический синдром (бледность кожных покровов, головокружение, учащение сердцебиения). Характерны носовые, десневые кровотечения, геморрагические высыпания на коже. Возникают боли в суставах, кожный зуд, боли в костях.
Пациент ощущает тяжесть и боли в левом подреберье вследствие выраженного увеличения селезенки. На фоне спленомегалии развивается синдром гиперспленизма, который заключается в массивном разрушении клеток крови (в основном эритроцитов) в синусоидах селезенки. В этом случае встречаются признаки гемолиза (желтушность кожи, слизистых оболочек, потемнение мочи).
Редкие симптомы связаны с необычной локализацией очагов экстрамедуллярного кроветворения – в легких (кашель, затруднение дыхания, кровохарканье), желудочно-кишечном тракте (боли в животе, кровавая диарея). При расположении очагов в центральной и периферической нервной системе наблюдаются эпилептические судороги, нарушения чувствительности, слабость движений в конечностях, вплоть до полного паралича.
Осложнения
При миелофиброзе часто образуются тромбы, которые приводят к острому нарушению мозгового кровообращения, инфаркту миокарда, тромбоэмболии легочной артерии. Стойкое снижение уровня лейкоцитов нередко сопряжено с различными инфекциями, приобретающими тяжелое течение. Наиболее неблагоприятным осложнением считается трансформация миелофиброза в миелолейкоз (бластный криз), трудно поддающийся терапии. К нетипичным осложнениям следует отнести патологические переломы из-за деструкции трубчатых костей и портальную гипертензию, причиной которой служит длительная обструкция микротромбами внутрипеченочных вен.
Диагностика
Курацией пациентов с миелофиброзом занимаются врачи-гематологи. При общем осмотре обращает на себя внимание изменение цвета кожных покровов, слизистых (бледность или желтушность), спленомегалия при пальпации и перкуссии селезенки, иногда достигающей гигантских размеров (до лобкового симфиза). Дополнительные методы диагностики включают:
- Общие лабораторные исследования. В начале заболевания в общем анализе крови выявляется увеличение эритроцитов, тромбоцитов, лейкоцитов, со временем сменяющееся на низкие показатели. Часто в периферической крови присутствуют незрелые формы эритроцитов, лейкоцитов (миелоциты, промиелоциты). В биохимическом анализе крови наблюдаются повышенные концентрации лактатдегидрогеназы (ЛДГ), ионизированного кальция. Отмечаются изменения коагулограммы – ускорение свертывания крови, уменьшение активированного частичного тромбопластинового времени, торможение процессов фибринолиза. В анализе мочи обнаруживаются уробилин, гемоглобин, ураты (соли мочевой кислоты).
- Исследование костного мозга. Образец костного мозга получают с помощью трепанобиопсии. Гистологическая картина зависит от фазы заболевания. Для ранней (префибротической фазы) характерны гиперплазия всех ростков кроветворения (гранулоцитарного, мегакариоцитарного, эритроидного) с незрелостью клеток. В позднюю (фибротическую) фазу определяется большое количество коллагеновых и ретикулярных волокон (фиброз), замещающих гемопоэтическую ткань, выраженная клеточная атипия. Высокий уровень бластных клеток (более 20%) свидетельствует о трансформации миелофиброза в острый лейкоз.
- Молекулярно-генетические тесты. Диагностика мутации генов JAK2V617F, CALR, MPL осуществляется методом FISH. Для идентификации аллельной нагрузки мутации проводится полимеразная цепная реакция real-time. Также выполняется HLA-типирование для решения вопроса о возможности трансплантации костного мозга.
- Цитогенетические и цитохимические анализы. При цитогенетическом исследовании (кариотипировании) клеток костного мозга находят аномалии 1, 3, 6 хромосом (транслокация, трисомия, комплексные нарушения). При анализе химического состава (цитохимии) нейтрофилов активность щелочной фосфатазы оказывается в 3 раза выше нормы.
Для достоверной постановки диагноза гематологическим сообществом разработаны специальные критерии. Большие критерии включают повышенную клеточность костного мозга с ретикулярным и коллагеновым фиброзом, наличие мутаций генов JAK2V617F, MPL, CALR. К малым критериям относятся анемия, спленомегалия, лейкоэритробластоз (присутствие в крови незрелых форм лейкоцитов, эритроцитов), а также повышение лактатдегидрогеназы. Диагноз считается подтвержденным, если имеются 2 больших критерия или 1 большой и 3 малых критерия.
Миелофиброз следует дифференцировать в первую очередь с гематологическими заболеваниями, такими как аутоиммунные гемолитические анемии, гемобластозы (лейкозы, лимфомы). Сочетание спленомегалии с симптомами интоксикации (слабостью, субфебрилитетом, ночной потливостью) требует исключения туберкулеза, подострого инфекционного эндокардита.
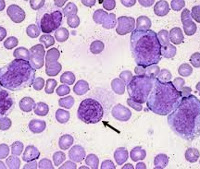
Незрелые формы эритроцитов (черная стрелка) и гранулоцитов (контурная стрелка) в периферической крови
Лечение миелофиброза
Прогноз и профилактика
Миелофиброз – это тяжелое заболевание с неблагоприятным прогнозом. С момента постановки диагноза средняя продолжительность жизни составляет около 5 лет. При манифестации в более молодом возрасте миелофиброз имеет менее агрессивное течение, что сопряжено с лучшим ответом на терапию и большей выживаемостью больных. Эффективных методов профилактики не разработано ввиду неизвестности этиологического фактора. Предупреждение развития вторичного миелофиброза заключается в своевременной диагностике и лечении патологий, на фоне которых он возникает - истинной полицитемии и эссенциальной тромбоцитемии.
2. Патофизиологические основы лечения сублейкемического миелоза. Патофизиология крови. Экстремальные состояния/ Под ред. А.И. Воробьева и Н.А. Горбуновой - 2004.
3. Критерии диагностики и современные методы лечения первичного миелофиброза/ Абдулкадыров К. М., Шуваев В. А., Мартынкевич И. С.// Вестник гематологии. - 2013 - №9(3).
4. Клинические рекомендации по диагностике и терапии Ph-негативных миелопролиферативных заболеваний (истинная полицитемия, эссенциальная тромбоцитопения, первичный миелофиброз. - 2014.
Читайте также: